Gene Therapy
Genes, which are carried on chromosomes, are the basic physical and functional units of heredity.
Genes are specific sequences of bases that encode instructions on how to make proteins.
Although genes get a lot of attention, it’s the proteins that perform most life functions and even make up the majority of cellular structures. When genes are
altered so that the encoded proteins are unable to carry out their normal functions, genetic disorders can result.
Gene therapy is a technique for correcting defective genes responsible for disease development.
There are several approaches for correcting the faulty genes.
Defective Gene – Genetic Disorder
Gene is said to be defective if there is change in the inherited gene. It is clear that any heritable change in a gene is brought about by mutation. Mutation
can be defined as any change in the base sequence of the DNA.
The consequences of mutation is-
# premature termination in the growth of the peptide chain,
# synthesis of non-functional protein.
In either event, the absence of the normal protein can lead to a variety of clinical manifestations depending on the structural or enzymatic role that
normally plays in the cell. Such conditions range from mild disorders that require no treatment (e.g., color blindness) to life threatening disease
(e.g., hemophilia, cystic fibrosis). Over 45,000 human diseases have been identified related directly to the genetic disorders.
Treatment of Genetic Disorders-Gene therapy versus conventional therapy
Limitations of the conventional therapy
1. Therapy based on the replacement of the missing or defective protein is available for only a few of these disorders. Example: Factor VIII for hemophilia,
adenosine deaminizes for SCIS, transfusion for sickle cell disease.
2. Conventional therapies are only partially effective in ameliorating the manifestations of the disease and are accompanied by significant complications.
3. For most genetic disease, providing the missing protein in a therapeutic fashion is not feasible due to the complex and fragile nature of the protein and the
need to deliver the protein to a specific subcellular localization (i.e., cell surface expression, lysosomal localization, etc.).
4. Transplantation of the major affected organ has been done in some instances (e.g., bone marrow transplantation for sickle cell disease, liver transplantation
for hyperlipidemia), but this has also severe limitations of organ availability and adverse consequences arising from the immune suppression required to
prevent rejection of an allogenetic tissue.
Problems overcome by gene therapy
A. Providing a normal copy of the defective gene to the affected tissues would circumvent the problem of delivering complex proteins, as the protein could
be synthesized within the cells using the normal cellular pathways.
B. The limited number of tissues is affected by most inherited disorders; this greatly simplifies the requirements for effective gene therapy, since a functional
copy of the gene need to be provided only to those tissues that actually require it.
So gene therapy is generally applied to correct defect in only part of the body and thus targeting of the therapeutic gene to a specialized area is important in
gene therapy.
C. If the gene transfer can be targeted to the major affected organs, thus side effects arising from ectopic gene expression in nontargeted cells might be
avoided.
D. As with other pharmaceutical agents, cell-specific targeting has the advantage of decreasing the effective volume of distribution and the amount of gene
transfer agent needed.
Where is gene therapy applied?
1. The majority of gene therapy trials underway are for the treatment of acquired disorders such as AIDS, cancer, CVS and inherited disorder arising from
single gene defects, such as AD, LSD, etc.
2. In case of inherited disorder, the defective gene that cause the disorder is the subject of intervention and
3. In the case of acquired diseases, either a defective gene that contributes to the disorder or a gene that mediates an unrelated biochemical process may be
the basis for intervention.
Example: Treatment of HIV infection potentially could rely on the interruption of viral processes that contribute to the pathogenesis of AIDS, antisense
mRNA, a dominant negtaive mutant protein.
Basis of Gene Therapy
The ability to transfect genes into cells and to cause their expression is the basis of gene therapy. So gene medicines are generally based on gene expression
system that contains a therapeutic gene and a delivery gene. The success of gene therapy is largely dependent on the development of a vector or vehicle that
can selectively and efficiently deliver a gene to target cell with minimal toxicity.
Two major methods have been described for gene transfer:
1. Viral-mediated gene transfer: various viruses are used as carrier, e.g., retrovirus, adenovirus, adenoassociated virus, etc.
2. Non-viral-mediated gene transfer: various types of synthetic vectors are used as carrier,. e.g., cationic lipid, cationic polymer, etc.
Candidate Disease for Gene Therapy
Diseases wherein gene therapy has been focused upon include-
Genetic Disease
|
Genetic Disease
|
Target cells
|
1. Severe Combined immunodeficiency(SCID/ADA)
|
Adenosine deaminase
|
Bone marrow cell or T-cell
|
2. Hemophilia- A
- B
|
Factor VIII deficiency
Factor IX deficiency
|
Liver, fibroblast
or bone marrow
|
3. Cystic fibrosis
|
Loss of CFTR gene
|
Airways in lung
|
4. Familial hypercholesterimia
|
Def. LDL receptor
|
Liver
|
5. Hemoglobinopathy
Thalessimia
Sickel cell anemia
|
structural defect
of alpha/beta- globin gene
|
bone marrow
|
Defective Gene – Genetic Disorder:
Disease-Acquired
|
Defect
|
Target cells
|
6. Neurologic diseases: Alzheimer’s disease
Parkinson’s disease
|
APP gene alpha-Syn gene
|
Neuron/gilal
Neuron
|
7. Cardiovascular disease; Atheroscleorosis
|
HDL
|
Vascular endothelial cell
|
8. Infectious disease:
AIDS ; Hepatitis B
|
|
T-cell
Liver
|
Approaches for gene therapy
Different approaches have been tried for effective gene therapy.
They are:
1. Gene modification- a) Replacement therapy
b) Corrective gene therapy
2. Gene transfer- a) Physical (microinjection, electroporation, etc.)
b) Chemical (Liposome, polymer, etc.)
c) Biological (Viral and non-viral vector
3. Gene transfer in specific cell lines-
a) Somatic gene therapy
b) Germline gene therapy
4. Eugenic approach (gene insertion)
Gene modification
A) Replacement Therapy
In this therapy, a defective gene is inserted somewhere in the genome so that its product could replace that of a defective gene. This approach may be suitable
for recessive disorders which are marked by deficiency of an enzyme or others proteins. It may not be suitable for dominant disorders which are associated
with the production of an abnormal gene product.
B) Corrective Gene Therapy
In this therapy it requires replacement of a mutant gene or a part of it with a normal. This can be achieved by using recombinant DNA technology. Another
form of corrective therapy involves the suppression of a particular mutation by a transfer RNA that is introduced in a cell.
Gene Transfer:
Gene transfer can be brought about by-
A) Physical,
B) Chemical, and
C) Biological means.
A) Physical: Several physical approaches have been developed to enhance the efficiency of gene transfer via naked DNA. These physical approaches allow
DNA directly to penetrate cell membrane. For example: Electroporation, gene gun, etc.
B) Chemical: Synthetic chemicals have been developed for gene transfer efficiently.
Example: Cationic lipid, cationic polymer, etc.
C) Biological: Viruses have been developed as a logical tools for gene transfer, because they have evolved mechanisms to enter the cell. Example:
Adenovirus, retrovirus, etc.
Gene Transfer in specific cell lines:
Somatic Gene Therapy
Somatic gene therapy involves the insertion of genes into specific somatic cells. They can function during the life time of an individual and hence correct a
genetic disease. However, it presents a number of practical problems.
Germ Line Therapy
Germ line therapy involves injection or insertion of gene into germ cells, into fertilized eggs. Here,the inserted gene would be passed onto future generations
too.
Eugenic Approach (gene insertion)
It is brought about by inserting genes to alter or improve complex traits of a person. For example: intelligence. However, it is far beyond the current
ethnological feasibility.
Essential Prerequisites for Gene Therapy
Before the patient is being subjected to gene therapy, several essential prerequisites are to be fulfilled. They are:
1. It must be possible to isolate the appropriate gene and to define its major regulatory regions.
2. Identification and harvesting of appropriate target cells.
3. Development of safe and efficient vectors with which the new gene could be introduced.
4. Clear evidence of experimental data on the adequate functioning of the inserted gene, life span of the recipient cell and that no untoward effect exists,
should be ensured.
5. Last but not the least, the patient or their family must be fully counseled.
Qualities of an ideal delivery system
An ideal delivery system would be one-
1. Which could accommodate a broad size range of inserted DNA?
2. Which could be targeted to specific types of cells?
3. Which would not permit replication of the DNA?
4. Which could provide long-term gene expression?
5. Which could be easily produced?
6. Which will be non-toxic and non-immunogenic?
Still such a delivery system does not exist.
There are two major delivery systems for gene transfer:
1. Viral-mediated gene delivery: various viruses are used as carrier,
e.g., retrovirus, adenovirus, adenoassociated virus, etc.
2. Non-viral-mediated gene delivery : various types of synthetic vectors are used as carrier,
e.g. cationic lipid, cationic polymer, etc.
Viral Mediated Gene Delivery
Why virus is used as a tool for gene delivery?
1. Viruses have evolved mechanisms to enter into the cell.
2. They can replicate inside the cell,
3. They use the cellular machinery to express their genes.
The natural life cycle of mammalian viruses has made them a vehicle for the transfer of therapeutic gene.
But for viral vectors to be useful, several viral functions must be altered-
1. At a minimum, the virus must be rendered replication-incompetent to prevent uncontrolled spread of the transgene,
2. Some elements of its own genome must be removed to allow for insertion of the transgene.
3. Beyond this, additional modifications are dependent on the specific virus to avoid its toxicity.
Retrovirus
Retroviruses are composed of an RNA genome that is packaged in an envelope derived from cell membrane and viral proteins. Retroviruses were first
described for gene transfer applications in 1981 and first utilized in clinical trials in 1989.
Retroviruses have had the greatest clinical use so far and offer the potential for long-term gene expression from a stably integrated transgene.
Characteristically, it offers a number of advantages in effective gene delivery to the cell:
1. It provides for official entry of genetic materials into a wide variety of cells,
2. It is well understood with simple molecular biology,
3. It integrates into host genome,
4. It has a potential control over the range of cells to be infected,
5. It is capable of gene expression,
6. It could carry up to 8Kb of coding information,
7. Above all, it establishes one way, nonreplicative infection of target cells,
8. It lacks irrelevant and potentially immunogenic proteins.
Drawbacks in use of retrovirus:
1. Retrovirus is limited to dividing cells,
2. Large scale production is technically possible, although purification and concentration potentially are problematic due to the instability of the virus.
Based on genomic structure, retroviruses are of two types:
1. Simple retrovirus, eg, most oncoviruses.
2. Complex retrovirus, e.g., lentivirus, spumavirus.
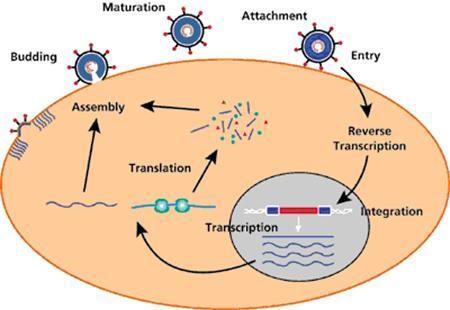
Replication Cycle of Retroviruses
Genome Structure of Retroviruses
Retrovirus
Figure 1. Proviral genome structure of the Murine Leukemia Virus (MLV), a simple retrovirus. Indicated are the 5’ and 3’ long terminal repeat (LTR; open boxes) regions comprising U3, R and
U5, as well as open reading frames (filled boxes) for gag, pol and envelope (env) proteins. Processed protein subunits are indicated in bold. att, attachment site; cap, 5’RNA capping site; pA,
polyadenylation site; PBS, primer binding site; SD, splice donor; y, packaging signal; SA, splice acceptor; PPT, polypurine tract; MA, matrix; CA, capsid; NC, nucleocapsid; PR, protease; RT,
reverse transcriptase; IN, integrase; SU, surface; TM, trans-membrane; E, enhancer; P, promoter.
(Source: http://enni82.hubpages.com/hub/Overview-of-Retrovirus-Life-Cycle# )
Genome Structure of Retroviruses
Essential characteristics of a simple retrovirus:
1. The viral DNA contains large redundant sequences at the two ends of the genome designated long terminal repeats (LTRs). LTRs can be further divided
into U3 (unique 3), R (repeat), and U5 (unique 5) regions.
U3: The viral promoters and transcriptional enhancers are located in the U3 region;
R: The R region is essential for reverse transcription and replication of all retroviruses.
In addition, the R regions of some viruses also contain elements important for gene expression.
U5: The U5 region contains sequences that facilitate the initiation of reverse transcription.
2. Immediately downstream of the 5’ LTR is a primer binding site (PBS) that has sequence complementarity to a portion of a cellular tRNA. Different
tRNAs are used by different viruses as primers for the initiation of reverse transcription.
3. The packaging signal (C) or encapsidation signal (E) are sequences that interact with the viral proteins to accomplish specific packaging of the viral RNA.
4. The coding regions of all retroviruses contain at least three genes.
Gag: The gag gene near the 5" end of the viral genome codes for Gag polyproteins that make up the viral capsid.
pol: The pol gene encodes reverse transcriptase and integrase. Reverse transcriptase copies the viral RNA to generate the viral DNA, whereas integrase
integrates the viral DNA into the host chromosome to form a provirus.
env: The env gene codes for the envelope polyprotein, which is cleaved into the transmembrane domain and the surface domain (SU).
The sequences that encode the viral protease (Pro) are always located between gag and pol and are most often expressed as either a part of the Gag
polyprotein or as a part of the Gag-Pol polyprotein.
5. The region between env and the 3’ LTR contains a purine-rich region known as polypurine tract (PPT) that is important for reverse transcription.
6. Short sequences at the two ends of the LTR are important for integration and are referred to as attachment sites (att). The att interact with integrase and are
necessary for efficient integration of the viral DNA.
Development of Retroviral Vector
●The first retroviral vector systems were derived from murine leukemia virus (MLV).
●There were (and are still) a number of reasons for choosing MLV as the basis for such gene delivery systems, including
the biology of this retrovirus is particularly well understood,
the MLV genome was among the earliest retroviral genomes molecularly cloned
and (iii) these viruses are able to infect cells efficiently.
●Retroviral vector systems consist of two components:
(i) a vector construct that carries the gene to be delivered and provides the genome for the recombinant virus,
and (ii) a cell line that provides the viral proteins required to produce the recombinant virus, known as packaging cells.
Construction of vector
►Retroviral vectors are constructed from the proviral form of the virus. The gaga, pol, and env genes are removed by the selectable marker to make room
for the gene(s) of therapeutic interest and to eliminate the replicative functions of the virus. Upto 8 Kilobases of heterologous DNA can be incorporated into
the retroviral vector.
►Along with the gene of therapeutic interest, gene encoding antibiotic resistance often are included in the recombibant as a means of selecting the
virus-harbouring cultured cells.
For example, aminoglysoside-3’-phosphotransferase gene for kanamycin, hygromycin B transferase gene for hygromycin. The antibiotic resistance gene
confers resistance to antibiotics.
►Sequences containing promoter and enhancer functions may also be included with the transgene to facilitate the efficient expression, and in some
circumstances, to provide for tissue-specific expression after administration in vivo. Alternatively, the promoter and enhancer functions contained in the
LTR
of the virus may be used for this purpose.
Marker gene Therapeutic gene with separate internal promoter to drive high level expression
Packaging Cell Lines or Helper Cell
To produce recombinant retroviral vector virions, the vector construct carrying the gene(s) to be delivered is introduced by physical gene transfer methods
(such as transfection, electroporation etc.) into a retroviral packaging cell line or helper cell. Helper cells are engineered culture cells expressing viral proteins
needed to propagate retroviral vectors; this is generally achieved by transfecting plasmids expressing viral proteins into culture cells. Most helper cell lines
are derived from cell clones to ensure uniformity in supporting retroviral vector replication.
These helper or packaging cells produce the viral structural (Gag and Env) proteins and enzymes (pol-encoded RT, IN), but are not able to package the
viralRNA encoding these proteins since the region required for encapsidation has been deleted. Instead the proteins recognise and associate with genomic
length RNA from the introduced vector construct, which carries an intact region, to form recombinant virus particles. The recombinant virus particles
carrying the retroviral vector genome bud out of the packaging cell line into the cell culture medium.
The virus-containing medium is either directly filtered to remove cells and cellular debris or then used to infect the target cell, or virus is purified and
concentrated before infecting target cells.
After the virus has bound to the receptor on the cell surface, the viral capsid is delivered into the cell and the viral RNA is reverse transcribed into a DNA
form which integrates into the host cell DNA. The integrated viral DNA (provirus) functions essentially as any other cellular gene and directs the synthesis
of the products of the delivered gene(s).
Major Problem with Retroviral Vector
The major problem with two-component retroviral vector systems arises as a result of the naturally occurring phenomenon of homologous recombination.
If the vector provirus and the provirus providing the structural proteins in the packaging cells recombine, there is a possibility that replication- competent
retrovirus will arise. Such virus is essentially a wild-type retrovirus and no longer carries the delivered gene(s).
Replication-competent virus rapidly infects many cells and may eventually cause insertional mutagenesis. Consequently, considerable effort has been
devoted to the design of superior packaging systems that drastically reduce the possibility of recombination occurring, as well as to produce improved, safer
vectors that cannot replicate even if recombination occurs.
Improvements to Packaging Cells
1. Improvements to packaging cells have involved removing as much of the retroviral information as possible to reduce the possibility of homologous
recombination occurring. The retroviral promoter and termination sequences can be replaced by heterologous promoters and termination sequences.
This has the additional advantage of allowing the use of promoters that are more strongly active than the retroviral promoter, thereby giving rise to higher
levels of viral protein production.
2. The coding information for the viral proteins cannot be removed by necessity, but these proteins can be made from separate constructs so that additional
recombination events are required to recreate a complete replication-competent retrovirus. This has been achieved by expressing the Gag and Pol proteins
from one construct and the Env proteins from a second construct.
3. In addition to the improvements to packaging cells, safer retroviral vector constructs also have been produced that carry an artificially inserted stop codon
in the Gag reading frame.
This ensures that even if replication-competent virus is generated, it will not be able to express its Gag and Pol proteins and thus virus assembly and release
will be inhibited.
Clinical Administration of Retrovirus
The clinical administration of retroviruses has been accomplished by:
● - ►Ex vivo transduction of patient’s cell,
● - ►Direct injection of virus into tissue,
● - ►Administration of retroviral producer cells.
1. Ex vivo gene transfer: The ex vivo approach has been most widely employed in human clinical trials. Although cumbersome in that it requires the
isolation and maintenance in tissue culture of the patients’ cell. It has the advantage that the extent of gene transfer can be quantified readily and a specific
population of cells can be targeted.
2. In vivo gene transfer: Retroviruses are being tested as potential agents to treat brain tumors which, in many circumstances, are relatively inaccessible.
Although the direct stereotactic injection of recombinant retrovirus into the target tissue is possible, the efficiency of gene transfer is very low.
Clinical Application of Retrovirus
Gene therapy approaches involving retroviral vectors can be used to treat several different types of human diseases. Retroviral vectors are being tested in
many clinical trials. A few examples of gene therapy clinical trials involving retroviral vectors are briefly described below:
1. The first clinical trials of human gene therapy using retroviral vector was designed to correct a genetic disorder known as adenosine deaminase (ADA)
deficiency. The patients lack adenosine deaminase which results in SCID. The lymphocytes of the patients were harvested and transduced or transfected
ex-vivo with a retrovirus containing a functional ADA gene and then the lymphocytes were reinfused into the host.
2. Hepatic genetic deficiencies have been experimentally treated with genes introduced via retroviral vectors. A case of hypercholesterolemia in rabbits have
been treated ex vivo and reimplantation of hepatocytes with the LDL-receptor gene.
Structure of Adenovirus
Fig. 1. Structure of adenovirus. The locations of the capsid and cement components are reasonably well defined. In contrast, the disposition of the core components and the
virus DNA is largely conjectural. (Source:http://vir.sgmjournals.org/content/81/11/2573.full)
Basic Characteristics of Adenovirus
►1. Adenovirus is a DNA virus having 60-90 nm in diameter. It has double stranded DNA genome and do not possess a lipid envelope
►2. Unlike retrovirus, adenoviruses have a characteristic morphology, with an icosahedral capsid consisting of three major proteins, hexon (II), penton base
(III) and a knobbed fibre (IV), along with a number of other minor proteins, VI, VIII, IX, IIIa and IVa2. The virus genome is a linear, double-stranded DNA
with a terminal protein (TP) attached covalently to the 5´ termini, which have inverted terminal repeats (ITRs). The virus DNA is intimately associated with
the highly basic protein VII and a small peptide termed mu. Another protein, V, is packaged with this DNA–protein complex and appears to provide a
structural link to the capsid via protein VI. The virus also contains a virus-encoded protease (Pr), which is necessary for processing of some of the structural
proteins to produce mature infectious virus.
►3. Adenoviral genome encodes approximately 15 proteins.
►4. Adenovirus infects a wide variety of human tissues such as respiratory epithelium, vascular endothelium, cardiac and skeletal muscle, peripheral and
central nervous system, hepatocytes, the exocrine pancreas and many tumor types.
►5. Adenovirus deliver their genomes to the nucleus and can replicate with high efficiency, they are prime candidates for the expression and delivery of
therapeutic genes.
Adenovirus-Mediated Infection of Target Cells
►Infection takes place when the fiber protein of capsid binds a cell surface receptor.
►Subsequently, peptide sequences in the penton base portion of the capsid engage integrin receptor domains (alpha3, beta3; alpha3, beta5) on the cell surface.
This leads to virus internalization via endosomal pathway where the viral particles begin to disassemble.
►The virus escapes the endosome prior to its fusion with lysosomal compartment and thus avoid digestion.
►The viral DNA is able to enter the target cell nucleus and begin transcription of viral mRNA.
Genomic Organization of Human Adenoviruses

FIGURE 1. Simplified transcription map of the Ad5 genome.
Essential characteristics of adenoviruses
1. Most Ad vectors are based on the well-characterized human Ad serotypes 2 or 5. The genome consists of a 36 kb linear, double-stranded DNA molecule.
Each end of the genome has an inverted terminal repeat (ITR) of 100-140 bp to which the terminal protein is covalently linked.
ITRs are required for viral DNA replication.
2. At 200 nucleotides of the 5Ꞌ extreme is located packaging signal (Y), sequence that directs the packaging of the viral genome through its interaction with
various viral and cellular proteins.
Gene are encoded on both strands of the DNA in a series of overlapping transcription units.
3. The viral genome is classified based on the timing of their expression (Fig. 1). Early genes (E1, E2, E3, and E4) are expressed before the onset of DNA
replication. Proteins encoded by the early genes function to activate other Ad genes, replicate the viral DNA, interfere with immune recognition of infected
cells, and modify the host-cell environment to make it more conducive to viral replication. The late genes (L1-L5) are expressed after DNA replication and
primarily encode proteins involved in capsid production and packaging of the Ad genome.
E1 = E1A genes activate the cascade of all other viral genes because only this gene needs the presence of cellular factors for its transcription. Furthermore,
E1A proteins inhibit cell replication, which contributes to viral genome replication more efficiently. The region E1B codies for proteins that inhibit apoptosis
and prepare the intranuclear environment for adenoviral replication (E1B 55K and E1B 19K).
E2= The E2 region encodes proteins required for viral DNA replication, including a single stranded DNA binding protein (E2a) and both the viral DNA
polymerase and the 55 kDa terminal protein (E2b).
E3= The E3 region encodes proteins that prevent the cellular immune response and thus, the adenovirus gets the time required to complete the infection
cycle.
E4= the E4 region encodes 7 open reading frames (ORFs) with clearly different functions. These functions include participation in the viral genome
replication, splicing, mRNA transport, inhibition of cellular protein synthesis, regulation of apoptosis and cell lysis.
Expression of the early genes leads to DNA replication, approximately eight hours after infection, and subsequent activation of the late genes under the
transcriptional control of the major late promoter, production of virus progeny, and finally, death of the host cell and virus release.
Design and Construction of Replication defective Human Adenoviral Vectors
Replication-deficient adenoviral vectors, similar to other viral vectors, are composed of the virion structure surrounding a modified viral genome. To date,
most vector particles are based on the
wild-type capsid structure which, in addition to protecting the viral DNA, provides the means to bind and enter (transduce) target cells. However, the viral
genome has been modified substantially.
These changes are designed to disable growth of the virus in target cells, by deleting viral functions critical to the regulation of DNA replication and viral
gene expression, while maintaining the ability to grow in available packaging or helper cells. Deletion of such sequences provides space within the viral
genome for insertion of exogenous DNA that encodes and enables appropriate expression of the gene of interest (transgene).
The subgroup C adenoviruses, serotypes 2 and 5 (Ad2 and Ad5), are among the best studied adenoviruses, and the viruses used most commonly as gene
transfer vectors. The vast majority of adenoviral vectors for gene therapy are E1 replacement vectors, where the transgene is inserted in place of the E1 region.
This E1 region deletion includes the entire E1a gene and approximately 60% of the E1b gene.
The vectors retain the immediate 5ꞌ end of the viral genome, including the left inverted terminal repeat (ITR) and encapsidation signal (Y), sequences required
for packaging, and the overlapping E1 enhancer region, in addition to the remainder of the viral genome (Figure 5.1). As the E1 gene products lead to
sequential activation of the major transcription units, deletion of this region greatly reduces early and late gene expression and renders the virus severely
replication impaired.
To provide more space within the adenoviral vectors for insertion of the transgene, the E3 region, not required for viral replication or growth, is also frequently
deleted. Occasionally, the transgene is inserted into this E3 region deletion. Adenoviral vectors lacking only E1 and E3 regions are referred to as first
generation, or Av1, vectors. Adenoviruses can effectively package DNA up to 105% of the genome size, allowing the accommodation of up to 8 kb of
exogenous DNA in E1/E3 deleted Av1 vectors.
Improvement of Transgene Expression
The transgene transcriptional unit consists of the elements required to enable appropriate expression of the transgene such as the promoter, the gene of
interest, and a polyadenylation signal, and, in most instances, is designed to maximize the expression of the exogenous gene. A large variety of promoters
have been utilized for transgene expression, the choice of which depends on the application and the target tissue.
Strong, constitutively expressed viral promoters such as the adenovirus major later promoter, the Rous sarcoma virus promoter, the cytomegalovirus (CMV)
promoter and a hybrid CMV enhancer/-actin promoter have been incorporated into recombinant adenoviral vectors. More recently, the use of cellular,
tissue-specific promoters such as the liver-specific albumin promoter, lung-specific cystic fibrosis transmembrane conductance regulator promoter,
the cardiac muscle-specific myosin light chain-2 promoter, and the hepatomaspecific -fetoprotein promoter has been described.
Finally, regulatable promoters responsive to hormonal or pharmacological agents have been incorporated into adenoviral vectors. The inclusion of
tissue-specific and/or regulatable promoters to the transgene expression cassette avoids the unknown consequences of over expression of genes in tissues
other than the targeted organ, and may, therefore, increase the safety of such vectors. An additional approach shown to increase the potency of the transgene
in adenoviral vectors is the introduction of genomic elements into the expression cassette.
For example, the addition of an intron to the human factor VIII (FVIII) cDNA boosted in vivo expression approximately 10-fold, and the inclusion of the
human factor IX (FIX) truncated first intron and 5 and 3 untranslated regions to the human FIX cDNA functioned synergistically to increase human
FIX plasma levels in transduced mice approximately 2000-fold. Finally, a variety of signals have been used to direct polyadenylation such as the simian
virus 40 polyadenylation signal.
Propagation and Purification of Adenoviral Vector
The propagation of Av1 adenoviral vectors, rendered almost completely replication defective by the deletion of the E1 region, requires the generation of cell
lines to complement the E1 functions in trans. Several human cell lines that constitutively express the E1 proteins have been established. To date, the most
widely used cell line, 293, consists of human embryonic kidney cells transformed with sheared Ad5 DNA that express the left 11% of the Ad5 genome.
While 293 cells allow replication of Av1 vectors to high titers, this cell line is not ideal for large-scale vector production. Recombination between homologous
E1 region sequences encoded in the vectors with those inserted in the 293 cell genome has the potential to generate replication-competent adenoviruses
(RCA). Furthermore, the presence of RCA in preparations of adenoviral vectors was shown to induce significant tissue damage in vivo.
The generation of RCA may be prevented by elimination of sequence homology between the vector DNA and the adenovirus sequences in the genome of the
complementing cells.
Unlike retroviral vectors, the stability of the adenovirus virion allows extensive purification and concentration without significant loss of activity. Procedures
for adenoviral vector purification involve harvest and disruption of infected cells using multiple freeze and thaw cycles, or sonication, and removal of the cell
debris by centrifugation. Vector is purified and concentrated in one to three CsCl centrifugation steps, followed by dialysis or chromatography.
However, for large-scale manufacturing, chromatographic methods which avoid CsCl centrifugation are desirable. Vector concentration is determined
spectrophotometrically, to evaluate particle number, and biologically, to measure infectivity by gene transfer or plaque assay. Concentrated vector preparations
containing 1011 plaque forming units per milliliter can be obtained routinely.
Limitations and Improvements of Adenoviral Vectors
Although recombinant adenoviral vectors have become increasingly popular gene delivery vehicles, there are two major limitations that could hamper their
eventual use in human gene therapy.
First, the adenoviral vectors usually mediate a short-term gene expression;
Second, adenoviral vectors tend to elicit strong immune and inflammatory responses in vivo.
A single large dose of adenovirus can efficiently provoke production of neutralizing antibodies directed to the viral particle, which in turn would preclude or
reduce the efficiency of repeated systemic administration.
♦ Several approaches have been explored to circumvent the immunogenicity of adenoviral vectors by changing the vector designs. For instance, a new
generation of adenoviral vectors has been constructed with deletion of E1, E2 and E4 genes in order to avoid expression of immunogenic viral proteins in
host cells.
♦ Alternatively, constitutive expression of E3 gp19K protein in E1-deleted vector has provided encouraging results with more stable transgene expression in
the liver and lung of animal models. The function of gp19K is to inhibit the transport of major histocompatibility complex class I molecules to cell surface,
leading to the impairment of antigen-presenting cells’ function and reduced clearance of adenoviral infected cells by CTL immune responses.
♦The recently developed gutless adenoviral vectors, which have most or all adenoviral genes deleted, have shown significantly reduced immunogenicity and
prolonged expression of homologous transgenes in mice.
♦Many approaches are being developed to control host immune responses at the time of infection. One of them is to transiently block cell adhesion and
co-stimulatory molecules, such as CD40 ligand, in order to prevent both cytotoxic response and production of virus-specific neutralizing antibodies.
Immunomodulating cyto-kines, such as IL-10 and IL-12, were also used to disrupt the balanced Th activation towards either Th1 (cytotixic) or Th2 (humoral)
subset, thereby reducing antibody production and cellular immune response, respectively. Because TNF-alpha has been shown to play a key role in
adenovirus-induced immune response, inhibition of this pathway may offer a particularly promising prospect in overcoming host cell immune responses.
♦Manipulation of virus capsid components by genetic engineering may provide another alternative to circumvent pre-existing humoral response to the
commonly used adenovirus serotype 5. In fact, vector capsids displaying chimeric Ad5/Ad12 hexon monomers were shown to overcome neutralizing
antibodies in C57BL/7 mice primed with Ad5. Interestingly, such chimeric capsid may also change the binding affinity to host cells. For instance, chimeric
capsid Ad5/Ad7 exhibited an enhanced binding affinity for human lung epithelial cells but significantly diminished efficiency for liver-directed gene transfer.
IMPROVEMENT OF EXPRESSION LEVEL (এটি চলমান point এর under এ)
♦Long-term expression of transgenes is desirable for replacement gene therapy. Several strategies have been developed to address the drawback of
adenovirus-mediated transient gene expression. For example, a chimeric adenoviral-retroviral vector has been constructed in order to maintain transgenes
within actively dividing cells. This chimeric virus was shown to infect cells and produce recombinant retroviruses that can infect surrounding cells and
integrate into host chromosome. Similarly, a hybrid adenoviral/ adeno-associated virus (AAV) was engineered and shown to integrate the transgene at a
specific locus of human chromosome 19.
The major difference between the two types of chimeric viruses is that AdV/AAV vector may also maintain efficient and lasting transgene expression in
non-dividing cells.
♦The transgene transcriptional unit consists of the elements required to enable appropriate expression of the transgene such as the promoter, the gene of
interest, and a polyadenylation signal, and, in most instances, is designed to maximize the expression of the exogenous gene. A large variety of promoters
have been utilized for transgene expression, the choice of which depends on the application and the target tissue.
♦While adenoviral vectors can efficiently transfer genes into a broad spectrum of cell types, this wide tropism also represents an apparent drawback when
gene delivery to a specific tissue is needed. Moreover, the transgene expression mediated by adenoviral vectors may require
fine-tuned regulation for some, if not all, therapeutic applications. Currently, expression of most transgenes is driven by ubiquitous promoters of viral origin,
such as the immediate-early promoter from human cytomegalovirus (CMV) and the Rous sarcoma virus long terminal repeat (LTR).
Although these promoters provide high levels of transgene expression, it is not always desirable and specific enough for a wide variety of therapeutic
applications. In this respect, endogenous promoters have been used to restrict transfer gene expression to particular cell types at a physiologically relevant
level.
For instance, tissue-specific transgene expression was achieved when a transactivator was coupled with tissue-specific promoters such as the liver-specific
albumin promoter, lung-specific cystic fibrosis transmembrane conductance regulator promoter, the cardiac muscle-specific myosin light chain-2 promoter,
and the hepatomaspecific -fetoprotein promoter has been described.
♦An additional approach shown to increase the potency of the transgene in adenoviral vectors is the introduction of genomic elements into the expression
cassette.
For example, the addition of an intron to the human factor VIII (FVIII) cDNA boosted in vivo expression approximately 10-fold, and the inclusion of the
human factor IX (FIX) truncated first intron and 5 and 3 untranslated regions to the human FIX cDNA functioned synergistically to increase human FIX
plasma levels in transduced mice approximately 2000-fold. Finally, a variety of signals have been used to direct polyadenylation such as the simian virus
40 polyadenylation signal.
Formation of Replication-Competent Adenovirus
Recombination between homologous E1 region sequences encoded in the vectors with those inserted in the helper cell, 293 cell genome has the potential to
generate replication-competent adenoviruses (RCA).
Furthermore, the presence of RCA in preparations of adenoviral vectors was shown to induce significant tissue damage in vivo. The generation of RCA may
be prevented by elimination of sequence homology between the vector DNA and helper cell DNA.
Application of Adenoviral Vector
One of the major advantages of adenoviral vectors is that, for a wide variety of cell types, they provide more efficient gene transfer compared with other
gene delivery approaches. This is especially true for in vivo gene transfer. Recombinant adenoviruses can transfer genes into both proliferating and quiescent
cells.
One limitation of adenovirus-mediated gene expression is that it is transient, ranging from two weeks to a few months, largely because recombinant
adenoviruses are replication-deficient and do not integrate into the host genome.Thus, adenovirus vectors may not be suitable for long-term correction of
chronic disorders but should be adequate for therapeutic strategies that require high and transient gene expression.
Use of Adenoviral Vectors for Cancer Gene Therapy
In the majority of cancer gene therapy trials, adenoviral vectors have been administered in vivo, and have been used to transfer drug-sensitive genes
(such as the herpes virus thymidine kinase), immuno-modulators (such as IL-2, IL-12, FasL), melanoma tumor antigens (such as MART-1), or tumor
suppressor genes (such as p53). To date, many tumor types have been treated with adenoviral based gene transfer. These include melanoma, prostate cancer,
mesothelioma, pancreatic cancer, lung cancer, neuroblastoma, glioblastoma, etc.
Use of Adenoviral Vectors for Non-cancer Gene Therapy
Monogenic Diseases
Adenoviral vectors have also played an important role in gene transfer studies directed to several monogenic diseases. For example, cystic fibrosis
(transmembrane conductance regulator). Adenoviral vectors expressing CFTR have been used in phase I clinical studies on the biosafety and efficacy of gene
transfer.
Genetic and Metabolic Liver Diseases
Because adenoviral vectors have been shown to mediate efficient gene transfer to hepatocytes, adenoviral directed gene delivery has been pursued as
potential therapies for a wide variety of genetic and metabolic liver disorders, such as lysosomal storage diseases, glycogen storage diseases,
phenylketonuria, and Tay-Sachs disease.
Neurodegenerative Diseases
Recently, the efficacy of adenoviral vectors has been demonstrated in several models of neurodegenerative diseases including Parkinson’s disease (PD) and
motor neuron diseases. In rat PD models, adenoviral vectors expressing either tyrosine hydroxylase, superoxide dismutase or glial-derived neurotrophic
factor improved the survival and functional efficacy of dopaminergic cells.
Cardiovascular Diseases and Tissue Regeneration
At least in animal models, adenoviral vectors have also been shown to effectively transducer therapeutic genes for several cardiovascular diseases, such as
atherosclerosis, cerebral ischemia, familial hypercholesterolemia, hypertension, and cardiac arrhythmias.
Adenoassociated Virus
Basic Characteristics of Adenoassocited Virus:
1. Adeno-associated viruses, from the parvovirus family, are small nonenveloped viruses (20-25 nm) with a genome of single stranded DNA (ssDNA),
which is approximately 4.7 kb in size.
2. These viruses can insert genetic material at a specific site on chromosome 19 with near 100% certainty.
3. There are a few disadvantages to using AAV, including the small amount of DNA it can carry (low capacity) and the difficulty in producing it.
4. This type of virus is being used, however, because it is non-pathogenic (most people carry this harmless virus).
5. In contrast to adenoviruses, most people treated with AAV will not build an immune response to remove the virus and the cells that have been successfully
treated with it.
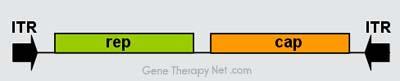
Genomic Organization of AAV Vectors
Figure 1. Genome organisation of Adeno-associated viruses.
The AAV genome is built of single-stranded deoxyribonucleic acid (ssDNA), either positive- or negative-sensed, which is about 4.7 kilobase long.
The genome comprises inverted terminal repeats (ITRs) at both ends of the DNA strand, and two open reading frames (ORFs): rep and cap (see figure 1).
The former is composed of four overlapping genes encoding Rep proteins required for the AAV life cycle, and the latter contains overlapping nucleotide
sequences of capsid proteins: VP1, VP2 and VP3, which interact together to form a capsid of an icosahedral symmetry.
Genomic Organization of AAV Vectors
♦The Inverted Terminal Repeat (ITR) sequences comprise 145 bases each. They were named so because of their symmetry, which was shown to be required
for efficient multiplication of the AAV genome.
Another property of these sequences is their ability to form a hairpin, which contributes to so-called self-priming that allows primase-independent synthesis
of the second DNA strand.
The ITRs were also shown to be required for both integration of the AAV DNA into the host cell genome and rescue from it, as well as for efficient
encapsidation of the AAV DNA combined with generation of a fully-assembled, deoxyribonuclease-resistant AAV particles.
♦With regard to gene therapy, ITRs seem to be the only sequences required in cis next to the therapeutic gene: structural (cap) and packaging (rep) genes can
be delivered in trans.
With this assumption many methods were established for efficient production of recombinant AAV (rAAV) vectors containing a reporter or therapeutic gene.
Application Of Adenoassociated Viral Vectors
1. Several trials with AAV are on-going or in preparation, mainly trying to treat muscle and eye diseases; the two tissues where the virus seems particularly
useful.
2. However, clinical trials have also been initiated where AAV vectors are used to deliver genes to the brain. This is possible because AAV viruses can infect
non-dividing (quiescent) cells, such as neurons in which their genomes are expressed for a long time.
3. In recent human trials, CD8+ immune cells have recognized the AAV infected cells as compromised and killed these cells accordingly.This action appears
to be triggered by part of the capsid or outer coat of the type 2 virus.
Limitations of Adenoassociated Viral Vectors
1. One of the major limitations for the use of AAV as a gene delivery vehicle is the relatively small packaging capacity. The unique ability of AAV vectors to
become joined into concatamers by head-to-tail recombination of the ITRs has been exploited as a means to increase the coding capacity. In this approach,
either the gene itself or the different elements of the transgene expression cassette are split over two AAV vectors that are administered simultaneously.
Transgene expression is obtained only after recombination between the two viral genomes, but the efficiency is often reduced as compared to single vector
transduction.
2. The AAV vectors do not contain any viral coding regions, and therefore, there is no toxicity associated with gene expression. However, a single injection
of AAV vector elicits a strong humoral immune response against the viral capsid, which will interfere with re-administration of the vector. Furthermore,
natural infections have resulted in a high prevalence of circulating neutralizing antibodies against AAV in the majority of the population, which may inhibit
transduction.
[such as the liver-specific albumin promoter, lung-specific cystic fibrosis transmembrane conductance regulator promoter, the cardiac muscle-specific myosin light chain-2 promoter, and the
hepatomaspecific -fetoprotein promoter has been described.]
Herpes Simplex Viral Vectors
∆Herpes simplex viruses (HSV) belong to the subfamily of Alphaherpesvirinae. Herpes viruses consists of a relatively large linear DNA genome of
double-stranded DNA 150 kb in length, encased within an icosahedral protein cage called the capsid, which is wrapped in a lipid bilayer called the envelope.
The envelope is joined to the capsid by means of a tegument. This complete particle is known as the virion.
∆The genome of Herpes viruses encodes some 100-200 genes. These genes encode a variety of proteins involved in forming the capsid, tegument and
envelope of the virus, as well as controlling the replication and infectivity of the virus.
∆Herpes simplex virus 1 and 2 (HSV-1 and HSV-2) are two species of the herpes virus family, which cause infections in humans. An infection by a herpes
simplex virus is marked by watery blisters in the skin or mucous membranes of the mouth, lips or genitals. The genomes of HSV-1 and HSV-2 are complex,
and contain two unique regions called the long unique region (UL) and the short unique region (US). Of the 74 known ORFs, UL contains 56 viral genes,
whereas US contains only 12. Transcription of HSV genes is catalyzed by RNA polymerase II of the infected host. Immediate early genes, which encode
proteins that regulate the expression of early and late viral genes, are the first to be expressed following infection. Early gene expression follows, to allow the
synthesis of enzymes involved in DNA replication and the production of certain envelope glycoproteins. Expression of late genes occurs last, this group of
genes predominantly encode proteins that form the virion particle.
Application of Herpes Simplex Viral Vector
Herpes viruses are currently used as gene transfer vectors due to their specific advantages over other viral vectors. Among the unique features of HSV
derived vectors are the very high transgenic capacity of the virus particle allowing to carry long sequences of foreign DNA, the genetic complexity of the
virus genome, allowing to generate many different types of attenuated vectors possessing oncolytic activity, and the ability of HSV vectors to invade and
establish lifelong non-toxic latent infections in neurons from sensory ganglia from where transgenes can be strongly and long-term expressed.
Alpha Virus Viral Vector
Alphaviruses, like Sindbis Virus and Semliki Forest Virus, belong to the Togaviridae family of viruses. There are 27 alphaviruses, able to infect various
vertebrates such as humans, rodents, birds, and larger mammals such as horses as well as invertebrates. Alphaviruses particles are enveloped have a 70 nm
diameter, tend to be spherical and have a 40 nm isometric nucleocapsid.
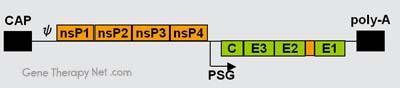
The genome of alphaviruses consists of a single stranded positive sense RNA. The total genome length ranges between 11 and 12 kb, and has a 5’ cap,
and 3’ poly-A tail. There are two open reading frames (ORF’s) in the genome, non-structural and structural. The first is non structural and encodes proteins
for transcription and replication of viral RNA, and the second encodes four structural proteins: Capsid protein C, Envelope glycoprotein E1, Envelope
glycoprotein E2, and Envelope glycoprotein E3. The expression of these proteins and replication of the viral genome all takes place in the cytoplasm of the
host cells.
Application of Alphavirus Viral Vector
1. Alphaviruses are of interest to gene therapy researchers, in particular the Ross River virus, Sindbis virus, Semliki Forest virus, and Venezuelan Equine
Encephalitis virus have all been used to develop viral vectors for gene delivery. Application of replication-deficient vectors leads to short-term expression,
which makes these vectors highly attractive for cancer gene therapy.
2. Alphavirus vectors carrying therapeutic or toxic genes used for intratumoral injections have demonstrated efficient tumor regression.
3. Of particular interest are the chimeric viruses that may be formed with alphaviral envelopes and retroviral capsids. Such chimeras are termed pseudotyped
viruses. Alphaviral envelope pseudotypes of retroviruses or lentiviruses are able to integrate the genes that they carry into the expansive range of potential
host cells that are recognized and infected by the alphaviral envelope proteins E2 and E1. The stable integration of viral genes is mediated by the retroviral
interiors of these vectors.
Limitation of Alphavirus Viral Vector
►There are limitations to the use of alphaviruses in the field of gene therapy due to their lack of targeting, however, through the introduction of variable
antibody domains in a non-conserved loop in the structure of E2, specific populations of cells have been targeted.
► Furthermore, the use of whole alphaviruses for gene therapy is of limited efficacy both because several internal alphaviral proteins are involved in the
induction of apoptosis upon infection and also because the alphaviral capsid mediates only the transient introduction of mRNA into host cells. Neither of
these limitations extends to alphaviral envelope pseudotypes of retroviruses or lentiviruses.
►However, the expression of Sindbis virus envelopes may lead to apoptosis, and their introduction into host cells upon infection by Sindbis virus envelope
pseudotyped retroviruses may also lead to cell death. The toxicity of Sindbis viral envelopes may be the cause of the very low production titers realized from
packaging cells constructed to produce Sindbis pseudotypes.
Non- Viral Vectors
An alternative to the use of viral vectors for gene delivery is to deliver genetic material in the form of bacterial plasmid DNA. In the simplest form, naked
plasmid DNA can be injected into skeletal muscle leading to transfection of muscle fibers close to the site of delivery. Though the transfection efficiency by
nonviral vectors is relatively lower than that by viral vectors, synthetic nonviral vectors are designed to overcome many of the problems associated with viral
vectors, such as risk of generating the infectious form or inducing tumorigenic mutations, risk of immune reaction, limitation to the size of genes incorporated
, and difficulty for the production to scale up.
Advantages
The advantages of nonviral carriers over their viral counterparts are:
(1) They are easy to prepare and to scale-up;
(2) They are generally safer in vivo;
(3) They do not elicit a specific immune response and can therefore be administered repeatedly;
(4) Nonviral vectors allow for the delivery of large DNA fragments and are also particularly suitable to deliver oligonucleotides to mammalian cells, which is
an excellent feature for the application of antisense strategies to downregulate the expression of certain genes; and (5) They are better for delivering cytokine
genes because they are less immunogenic than viral vectors.
Gene Transfer with Naked DNA
The simplest approach to nonviral delivery systems is direct gene transfer with naked plasmid DNA. Simple injection of plasmid DNA directly into a tissue
without additional help from either a chemical agent or a physical force is able to transfect cells. Local injection of plasmid DNA into the muscle, liver, or
skin, or airway instillation into the lungs, leads to low-level gene expression. Specific or nonspecific receptors on the cell surface that bind and internalize
DNA have been implicated as a mechanism.
Plasmid Design
☼The design and engineering of plasmids to obtain maximum transfection has been extensively researched. In addition to the transgene of interest, plasmid
DNA molecules typically contain several regulatory signals such as promoter and enhancer sequences that play an important role in regulating gene
expression. In viral delivery vectors, such signals can be endogenously present or artificially engineered in the virus genome. In addition, splicing and
polyadenylation sites are present in the transgene construct that help in the correct processing of the mRNA generated after transcription. Some vectors also
have introns that may increase premRNA processing and nuclear transport.
☼Promoter sequences play a vital role in initiating gene transcription. Promoter sequences offer recognition sites for the RNA polymerase to initiate the
transcription process. Higher efficiency can be obtained by engineering the plasmid with strong tissue- or tumor-specific promoters. Commonly used
promoter sequences are derived from viral origins such as cytomegalovirus (CMV) and roux sarcoma virus, or are obtained from human origins such as alpha
actin promoter. However, sometimes promoters can lose their activity upon immune stimulation.Promoter sequences may also play an important role in
determining the immune response of the cell to the gene product. For example, it was demonstrated that human muscle creatine kinase promoter has no
immunostimulatory effect in mice, as opposed to the commonly used CMV promoter sequence, during the expression of a gene vaccine encoding the hepatitis
B surface antigen.
☼Enhancers are regions in the plasmid DNA that enhance the production of the gene of interest by as much as several hundred times. Enhancers can be tissue
specific and can be present on the plasmid locus either upstream or downstream from the promoter region. Transcription efficiency can be substantially
improved by the choice of suitable enhancers. For example, promoters and enhancers derived from immunoglobulin genes have been used to increase gene
transduction in hematopoietic cells and to improve specificity of viral vectors useful in the treatment of hematological malignancies. Muscle creatine kinase
enhancer has been useful for enhanced targeted expression of transgenes for gene therapy to correct for muscular dystrophy.
Application of Naked DNA
Gene transfer with naked DNA is attractive to many researchers because of its simplicity and lack of toxicity. Practically, airway gene delivery and
intramuscular injection of naked DNA for the treatment of acute diseases and DNA-based immunization, respectively, are 2 areas that are likely to benefit
from naked DNA-mediated gene transfer, provided that further improvements are made in delivery efficiency and duration of transgene expression.
Limitations of Naked DNA and How to Overcome
◊ A broad application of naked DNA–mediated gene transfer to gene therapy may not be conceivable because DNA, being large in size and highly
hydrophilic, is efficiently kept out of the cells in a whole animal by several physical barriers. These include the blood endothelium, the interstitial matrices,
the mucus lining and specialized ciliate/tight junction of epithelial cells, and the plasma membrane of all cells. In addition, DNA degradation by intra- and
extracellular nuclease activities further reduces the chance that DNA entering nuclei will be intact and functional.
◊ The current strategy for improving naked DNA–based gene transfer is to include in DNA solution substances capable of enhancing the efficiency of DNA
internalization by target cells.
For example, transferrin has been shown to enhance transfection in vitro. The addition of water-immiscible solvents, non-ionic polymers, or surfactants, or
the use of hypotonic solution, has also been shown to elevate gene transfer across cell membranes. Also, several nuclease inhibitors have been shown to
enhance naked DNA–mediated gene transfer in cultured cells, muscle, and lungs.
Gene Transfer by Physical Methods
Physical approaches have been explored for gene transfer into cells in vitro and in vivo. Physical approaches induce transient injuries or defects on cell
membranes, so that DNA can enter the cells by diffusion. Gene delivery employing mechanical (particle bombardment or gene gun), electric (electroporation),
ultrasonic, hydrodynamic (hydrodynamic gene transfer), or laser-based energy has been explored in recent years.
Transfer by Gene Gun
Particle bombardment through a gene gun is an ideal method for gene transfer to skin, mucosa, or surgically exposed tissues within a confined area. DNA is
deposited on the surface of gold particles, which are then accelerated by pressurized gas and expelled onto cells or a tissue. The momentum allows the gold
particles to penetrate a few millimeters deep into a tissue and release DNA into cells on the path. This method has been successfully used to deliver genes into
liver, skin, pancreas, spleen and tumours. This method may be used for DNA-based immunization.

Above: Helios Gene Gun schematic (Above left and right images courtesy of BioRad)
(Source: http://www.bio.davidson.edu/courses/molbio/molstudents/spring2003/mcdonald/gene_gun.html)
Gene Transfer By Electroporation
It is one of the several physical methods used for gene transfer. In this process, DNA is transferred into cells in suspension by applying pulses of high voltage
electricity that created pores in cell membrane or increase the permeability of protoplast membrane thereby facilitating the entry of DNA molecules ino the
cells.
Electroporation is a versatile method that has been extensively tested in many types of tissues in vivo, among which skin and muscles are the most extensively
investigated, although the system should work in any tissues into which a pair of electrodes can be inserted.
Advantages of Electroporation
1. The level of reporter gene expression obtained was 2 to 3 orders of magnitude higher than that with plasmid DNA alone.
2. DNA as large as 100 kb has been effectively delivered into muscle cells.
Electroporation Process
Long-term expression over 1 year after a single electroporation treatment was seen.
Gene transfer by electroporation showed less variation in effi ciency across species than did direct DNA injection.
Limitations of Electroporation Process
Several major drawbacks exist for in vivo application of electroporation.
First, it has a limited effective range of ~1 cm between the electrodes, which makes it difficult to transfect cells in a large area of tissues.
Second, a surgical procedure is required to place the electrodes deep into the internal organs.
Third, high voltage applied to tissues can result in irreversible tissue damage as a result of thermal heating. Ca 2+ influx due to disruption of cell membranes
may induce tissue damage because of Ca 2+ -mediated protease activation.
The possibility that the high voltage applied to cells could affect the stability of genomic DNA is an additional safety concern. However, some of these
concerns may be resolvable by optimizing the design of electrodes, their spatial arrangement, the field strength, and the duration and frequency of electric
pulses.
Electroporator with square wave and exponential decay waveforms for in vitro, in vivo, adherent cell and 96 well electroporation applications. Manufactured by BTX
Harvard Apparatus, Holliston MA USA.
Cuvettes for electroporation. These are plastic with aluminium electrodes and a blue lid. They hold a maximum of 400 μl.


Electronic Pulse Delivery (EPD) Technology
EPD is a sophisticated method which subjects the target cells to a precisely controlled pulses of electrical field in a computer controlled molecular transfer
system. The EPD system consists of
●► –a controller,
and●► –a reaction chamber.
The controller, driven by an EPD computer accurately controls the output of electronic pulse, which in turn are controlled by different parameters.
The reaction chamber, where the DNA transfer takes place holds the target cells and therapeutic gene as a mixture. The target cells suitable for EPD delivery
include haematopoietic stem cells, hepatocyte, fibroblasts, and myoblasts.
Advantages of EPD Technology
1. Using this technology, large molecules can be transferred into target cells (e.g., DNA-15kb).
2. EPD inserts only the therapeutic genes into the target cells without the involvement of any other molecule.
3. It has no toxic effect on the target cells and is completely free from other potential hazards.
4. EPD technology has remarkable transfer efficiency (80-90%).
5. It can be operated in a batch or continuous process.
6. The time taken by this method for transfer is very short (from few seconds to a minute).
EPD technology has been confused with electroporation procedure. In electroporation, the target cells are damaged after which the molecules transfer takes
place. Furthermore, electroporation often applies a significant electric current and need no contact by electrode to the mixture of genes and the target cells.
Ultrasound-Facilitated Gene Transfer
Ultrasound can facilitate gene transfer at cellular and tissue levels. A 10- to 20-fold enhancement of reporter gene expression over that of naked DNA has
been achieved.
The transfection efficiency of this system is determined by several factors, including the frequency, the output strength of the ultrasound applied, the
duration of ultrasound treatment, and the amount of plasmid DNA used. The efficiency can be enhanced by the use of contrast agents or conditions that make
membranes more fluidic. The contrast agents are air-filled microbubbles that rapidly expand and shrink under ultrasound irritation, generating local shock
waves that transiently permeate the nearby cell membranes.
Unlike electroporation, which moves DNA along the electric field, ultrasound creates membrane pores and facilitates intracellular gene transfer through
passive diffusion of DNA across the membrane pores. Consequently, the size and local concentration of plasmid DNA play an important role in determining
the transfection efficiency. Efforts to reduce DNA size for gene transfer by methods of standard molecular biology or through proper formulation could result
in further improvement. Interestingly, significant enhancement has been reported in cell culture and in vivo when complexes of DNA and cationic lipids have
been used.
Since ultrasound can penetrate soft tissue and be applied to a specific area, it could become an ideal method for noninvasive gene transfer into cells of the
internal organs.
So far, the major problem for ultrasound-facilitated gene delivery is low gene delivery efficiency.
Gene Delivery by Chemical Methods
As has been discussed, gene expression can be achieved by direct intratissue injection of naked plasmid DNA, gene transfer via other routes of administration
such as intrathecal and intravenous injection will generally require the use of a delivery vector or vehicle.
Various types of synthetic vectors have been developed for gene transfer. Theses include:
1. Cationic lipids;
2. Cationic synthetic polymers;
3. Peptides that act in a nonspecific manner;
4. Peptide and carbohydrate based targeting ligand
Among these, cationic lipid and polymers have been studied most extensively. In this system, DNA is formulated into condensed particles of cationic lipid or
polymer. The DNA containing particles are subsequently taken up by cells via endocytosis in the form of intracellular vesicles, from which a small fraction of
the DNA is 
Cationic Lipid and Polymer
1. Cationic Lipid
In 1987, Felgner et al. first reported that a double chain monovalent quaternary ammonium lipid, N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium
chloride, effectively binds and delivers DNA to cultured cells, hundreds of new cationic lipids have been developed, differ by the number of charges in their
hydrophilic head group and by the detailed structure of their hydrophobic moiety. Cationic lipid, as the name indicates consists mainly of a positively charged
lipid, thus it has been used to reduce the net negative surface charge on DNA plasmid based gene expression system, in order to reduce charge-charge
repulsion at the surface of biological membranes. Such lipids form a stable complexes with the gene expression system.
Although some cationic lipids alone exhibit good transfection activity, they are often formulated with a noncharged phospholipid or cholesterol as a helper
lipid to form liposomes. For example, LIPOFECTIN, a novel and highly efficient DNA transfection system consisted of the positively charged quarternery
amino lipid, DOTMA in a 1:1 weight mixture with dioleyl-phosphatidyl choline (DOPE).
As DOPE can fuse with endosomal membrane, it is generally included to effect the endosomal release of a gene expression system. Spontaneous mixing
between cationic lipids and cellular lipids in the membrane of the endocytic vesicles is crucial to the endosome-releasing process.
Spontaneous lipid mixing in endosomes becomes more profound when a non-bilayer-forming lipid such as dioleoylphosphatidylethanolamine (DOPE) is used
as the helper lipid, rather than a bilayer-forming lipid, dioleoylphosphatidylcholine. Inclusion of DOPE is believed to increase membrane fluidity and
facilitate lipid exchange and membrane fusion between lipoplexes and the endosomal membrane. A high local concentration of DOPE, which has a strong
tendency to form an inverse hexagonal phase, may lead to a nonbilayer lipid structure and cause membrane perturbation and endosome destruction.
However, some multivalent lipids have intrinsic transfection activity, and a helper lipid does not have a major impact on overall transfection activity,
indicating that multivalent cationic lipids work on a different mechanism.
Upon mixing with cationic liposomes, plasmid DNA is condensed into small quasi-stable particles called lipoplexes. DNA in lipoplexes is well protected
from nuclease degradation. Lipoplexes are able to trigger cellular uptake and facilitate the release of DNA from the intracellular vesicles before reaching
destructive lysosomal compartments.
The transfection efficiency of lipoplexes is affected by:
1) The chemical structure of the cationic lipid,
(2) The charge ratio between the cationic lipid and the DNA,
(3) The structure and proportion of the helper lipid in the complexes,
(4) The size and structure of the liposomes,
(5) The total amount of the lipoplexes applied, and
(6) The cell type.
The first 4 factors determine the structure, charge property, and transfection activity of the lipoplexes. The remaining 2 define the overall toxicity to the
treated cells, and the susceptibility of the cells to a particular lipid-based transfection reagent.
Figure 6: Model of the intracellular delivery of plasmid mediated by cationic lipid in plasmid DNA-cationic lipid complexes (lipoplexes). Following binding
(step 1) and endocytosis (step 2) into a target cell, the lipoplexes are transferred to endosomal compartments (step 3).
The membrane of the lipoplex then fuses with the endosomal membrane due to the tendency of positively charged and negatively charged membranes to
adhere and fuse, leading to lipid mixing between lipoplex and endosomal lipids. Anionic lipids from the endosomal membrane then displace cationic lipids
from the plasmid DNA, leading to release of plasmid and further formation of non-bilayer structures (step 4).
Cationic Lipid-Undesirable Effect
►Cationic lipids as oligonucleotide carriers have several disadvantages. The main disadvantages of cationic lipids are their toxicity and markedly decreased
activity in the presence of serum.
►Once administered in vivo, lipoplexes tend to interact with negatively charged blood components and form large aggregates that could be absorbed onto
the surface of circulating red blood cells, trapped in a thick mucus layer, or embolized in microvasculatures, preventing them from reaching the intended
target cells in the distal location. Newer cationic lipid formulations are available that exhibit decreased toxicity.e.g. The inclusion of a helper lipid (DOPE or
cholesterol)in new formulation reduces the effective charge ratio required to deliver oligonucleotides into cells and permits delivery in the presence of high
serum concentration.
►Toxicity related to gene transfer by lipoplexes has been observed. Acute inflammation reactions have been reported in animals treated with airway
instillation or intravenous injection of lipoplexes. Symptoms include acute pulmonary hypotension, induction of inflammatory cytokines, tissue infiltration
of neutrophils in lungs, decrease in white cell counts, and in some cases tissue injury in liver and spleen. In humans, various degrees of adverse inflammatory
reactions, including flulike symptoms with fever and airway inflammation, were noted among subjects who received aerosolized GL67 liposomes alone or
lipoplexes. These early clinical data suggest that these lipoplex formulations are inadequate for use in humans.
Application of Cationic Lipid Based Gene Transfer
Despite these undesirable characteristics, lipoplexes have been used for in vivo gene delivery to the lungs by intravenous and airway administration.
Transgene expression was clearly detectable but in most cases was insufficient for a meaningful therapeutic outcome. For airway gene delivery to the lungs,
animal studies using lipoplexes prepared from 3 b -[N-(N′,N′-dimethylaminoethane) carbamoyl]cholesterol (DC-Chol) and
Cationic Polymer
DOPE have shown that this procedure was mild to the host and partially effective in correcting genetic defects in a cystic fibrosis transmembrane regulator
mutant model.
Polymeric gene carriers have been studied because of some advantanges over the lipid systems:
(1)Relatively small size and narrow distribution of complex.
(2) High stability against nucleases; and
(3) Easy control of physical factors (e.g. hydrophilicity and charge) by co-polymerization.
Like cationic lipid, positively charged polymer form complex by interacting with negatively charged DNA. The complex is called polyplex, which results
from the electrostatic attraction between the cationic charge on the polymer and the negatively charged DNA. These polyplexes effect efficient gene delivery
into a variety of cell types.
A cationic polymer/DNA complex may contain one or more plasmid DNA. The formation of cationic polymers/DNA complexes protects DNA from both
extracellular and intracellular degradation during the trasfection. For in vitro gene delivery, cell membrane is the first barrier. The complexes are taken up by
cells usually through endocytosis, and the route of uptake determines the subsequent DNA release, trafficking and lifetime in the cells. Endocytosis is a
multi-step process including binding, internalization, forming of endosomes, fusion with lysosomes and lysis.
Since DNA and associated complexes are easily degraded in endosomes and lysosomes due to the extremely low pH and enzymes, they must rapidly escape
from endosomes. Chloroquine is a weak base. The chloroquine added into the polymer/DNA complexes or cell culture medium raises the pH value of
endosomes (normal pH of endosome is 5.5) after uptake by cells, facilitates endosome escaping and improves the trasfection efficiency.
The transfection efficiency mediated by cationic polymers possessing high buffer capacity at pH 5 7, such as polyethylenimine (PEI) and polyamidoamine
dendrimer, is high due to their ability to capture the protons entering the endosomes during their acidification leading to swelling and destabilization of
endosomes. After escaping from endosomes, DNA or polymer/DNA complexes move through cytoplasm to the nucleus by diffusion [8].
Cell nuclear membrane is another potent tight barrier for foreign DNA, which isolates the nucleoplasm from cytoplasm in order to control important cell
activities such as DNA replication and transcription. The nuclear pores on the nuclear membrane have huge and complex structures. Molecules smaller than
300 kg/mol can pass through the nuclear pores by passive diffusion, whereas the larger molecules are generally transported by the active signal-mediated
process. DNA or polymer/DNA complexes enter the nucleus probably through the nucleus pores.
Over the past two decades, various natural and synthetic cationic polymers were applied in gene delivery[11] including polyethylenimine (PEI),
polyamidoamine dendrimer, chitosan, gelatin, cationic peptides, cationic polyesters (PAGA), cationic polyphosphoesters (PPE), poly(vinyl pyridine),
poly[(2-dimethylamino)ethyl methacrylate], etc. Among the polymer vectors currently used, PEI is the most successful one and has become the gold
standard of polymer gene delivery vector.

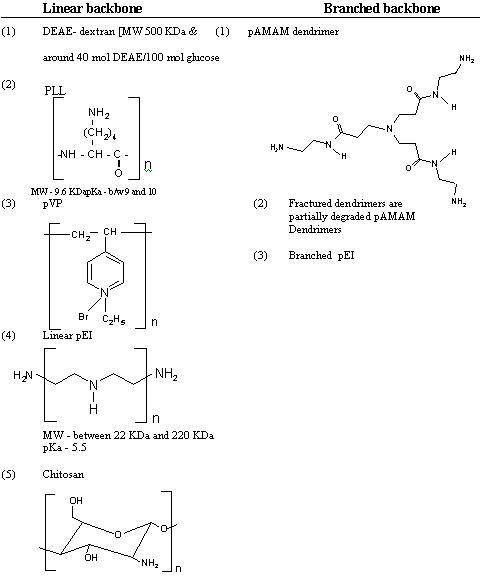
Oligonucleotides
Oligonucleotides are short single-stranded segments of DNA. In general, they are 13–25 nucleotides in length and hybridize to a unique sequence in the total
pool of targets present in cells. Due to their antigene and antisense application they are used either as therapeutic agents or as tools to study gene function.
Although it is not a complicated matter to synthesize phosphodiester oligonucleotides, their use is limited as they are rapidly degraded by the intracellular
endonucleases and exonucleases, usually via 3’-5’ activity. In addition, the degradation products of phosphodiester oligonucleotides, dNMP mononucleotides,
may be cytotoxic and also exert antiproliferative effects. Upon cellular internalization it can selectively inhibit the expression of a single protein.
Mechanism of action
Oligonucleotides are in theory designed to specifically modulate the transfer of the genetic information to protein, but the mechanisms by which an
oligonucleotide can induce a biological effect are subtle and complex. However, they produce the biological effects through two mechanisms, antigene and
antisense mechanisms.
For antigene applications, oligonucleotides must enter the cell nucleus, form a triplex with the double-stranded genomic DNA, and inhibit the translation as
well as the transcription processes of the protein.
On the basis of antisense mechanism of action, two classes of antisense oligonucleotide can be discerned:
(a) the RNase H-dependent oligonucleotides, which induce the degradation of mRNA; and (b) the steric-blocker oligonucleotides, which physically prevent
or inhibit the progression of splicing or the translational machinery.
The majority of the antisense drugs investigated in the clinic function via an RNase H-dependent mechanism. RNase H is a ubiquitous enzyme that
hydrolyzes the RNA strand of an RNA/DNA duplex. Oligonucleotide-assisted RNase H-dependent reduction of targeted RNA expression can be quite
efficient, reaching 80–95% down-regulation of protein and mRNA expression.
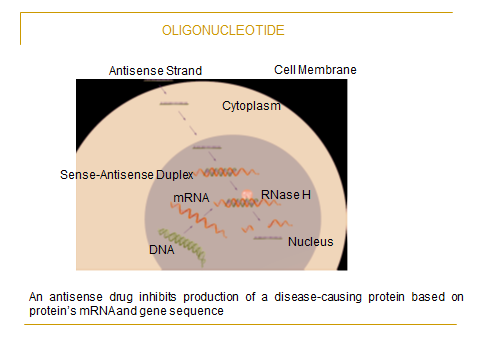
{RNase H-dependent antisense mechanism. Single-stranded oligonucleotides are transported across the plasma membrane, by either poorly characterized natural processes or by
the use of facilitators such as cationic lipids (step 1). Once in the cytoplasm, single-stranded oligonucleotides rapidly accumulate in the cell nucleus (steps 2 and 3), where they bind to their
targeted RNA (step 4). Once bound to the RNA, RNase H recognizes the oligonucleotide/RNA duplex as a substrate, cleaving the RNA strand and releasing the antisense oligonucleotide
(step 5). Although the cleavage of the RNA by RNase H is shown to occur in the nucleus, RNase H is also present in the cytosol, allowing for cleavage to occur in that cellular compartment
as well}
(Source: http://www.nature.com/onc/journal/v22/n56/fig_tab/1207231f1.html)
Furthermore, in contrast to the steric-blocker oligonucleotides, RNase H-dependent oligonucleotides can inhibit protein expression when targeted to virtually
any region of the mRNA. Whereas most steric-blocker oligonucleotides are efficient to bind to key sequences in the target which may inhibit the target’s
ability to interact with the cellular machinery required for protein synthesis. For example, oligonucleotide interaction with the initiation codon or with the
splicing sequences can cause transcriptional arrest by this route.
Design of Oligonucleotides
Design is critical for the clinical efficacy of oligonucleotides and should include consideration of length, chemistry, conformation, and ability to hybridize
with the target mRNA.
Length.
The suggested optimal length for oligonucleotides with efficient antisense activity ranges from 12 to 28 bases. There is a consensus that very short sequences
are likely to be nonspecific and sequences longer than 25 bases would experience difficulty in cellular uptake. As the length of the molecule increases,
hydrogen bonding between the base pairs and stacking interactions increases, leading to An increased overall affinity for the target.
However, longer oligonucleotides are inherently difficult to import into cells because of their size and tend to self-hybridize, thereby affecting their uptake.
Even in cell culture experiments, the optimum length suggested for oligonucleotides varies considerably.
Backbone Modifications
Oligonucleotides having the endogenous phosphodiester backbone are susceptible to degradation by nucleases and hence have limited use for antisense
applications. Various chemical modifications to the backbone have been used to improve oligonucleotide stability. The most common modifications include
the introduction of phosphorothioate and methyl phosphonate linkages in the backbone. Phosphorothioate analogs are chosen for their stability against
nucleases and the methylphosphonate backbone for its relative hydrophobicity and ease of diffusion across membranes. Phosphorothioate backbone
oligonucleotides can have significantly increased biological half-life compared to their corresponding unmodified phosphodiester oligonucleotides.
Backbone of Oligonucleotide
Hybridization Capacity
Antisense oligonucleotides are designed primarily for their ability to hybridize with the mRNA of interest. The mRNA has a complex secondary and tertiary
spatial structure that restricts the accessibility of certain segments of the molecule to hybridize with the oligonucleotide. Consequently, although it is
theoretically possible to design antisense constructs to virtually any part of the mRNA, not all oligonucleotides are efficient in inhibiting protein synthesis.
Complex algorithms predicting mRNA structure, “gene-walking,” RNase H mapping, and combinatorial screening are some of the strategies that are being
explored to predict hybridization-accessible sites on mRNA molecules.
Secondary Conformation
The presence of some specific sequences in the oligonucleotide can allow it to have a preferred conformation. For example, it was demonstrated that the
presence of continuous guanines at the 3'-end of an oligonucleotide made it resistant to degradation because of the ability of these guanines to assemble
into hyperstructures in vivo. These hyperstructures were resistant to nuclease activity and were efficiently taken up by cells. The secondary structure of
oligonucleotides can be affected by divalent cations such as Ca2+ and Mg2+ that typically occur in the cellular environment, thereby possibly affecting their
activity or cellular uptake.
Clinical Applications of Oligonucleotides
For therapeutic purposes, oligonucleotides can be used to selectively block the expression of proteins that are implicated in diseases. With successful
antisense inhibition of proteins in animal models, the first antisense drug, fomivirsen sodium (Vitravene, Isis Pharmaceuticals, Carlsbad,CA) was approved
for the treatment of cytomegalovirus retinitis in AIDS patients in 1998. Fomivirsen sodium is formulated as an intravitreal aqueous injection in sodium
bicarbonate buffer at pH 8.7.
Antisense oligonucleotides such as MG98 and ISIS 5132, designed to inhibit the biosynthesis of DNA methyltransferase and c-raf kinase, respectively, are
in human clinical trials for cancer. ISIS 2302, targeting ICAM-1, is being investigated for the treatment of ulcerative colitis.
Two other drug candidates, Affinitak and Alicaforsen (Isis Pharmaceuticals), are in phase 3 clinical trials for non-small cell lung cancer and Crohn’s disease,
respectively. Genasense (oblimersen sodium), developed by Aventis (Bridgewater, NJ) and Genta (Berkeley Heights, NJ), is being investigated in advanced
phase 2 trials in combination with other chemotherapy regimens for a range of cancers including malignant melanoma, chronic lymphocytic leukemia, and
multiple myeloma.
Vitravene Tm Injection (Fomivirisen sodium intravitreal injection)
Vitravene is a sterile, aqueous, preservative-free, bicarbonate buffered solution for intravitreal injection. It is represented by the following structural
formula-
5’-GCG TTT GCT CTT CTT CTT GCG-3’
So fomivirisen sodium is a phosphorotioate oligonucleoltie, twenty one nucleotides in length.
Vitrvene Injection
Indications
Vitravene is indicated for the local treatment of cytomegalovirus (CMV) retinitis in patients with AIDS, who are intolerant of or have a contraindication to
other treatment for CMV retnitis or who were insufficiently responsive to previous treatments for CMV retinitis.
Mechanism of action
Vitravene inhibits human cytomegalovirus (HCMV) replication through an antisense mechanism. The nucleotide sequence of fomivirisen is complementary
to a sequence in mRNA transcripts of the major immediate early region 2 (IE2) of HCMV. This region of mRNA encdes several proteins responsible for
regulation of viral gene expression that are essential for production of infectious CMV.
Binding of fomivirisen to the target mRNA, results in inhibition of IE2 protein synthesis, subsequently inhibiting virus replication.
siRNA (Small Interfering RNA)
A number of technologies have been used in an attempt to mediate the down-regulation of gene expression. For example, anti-sense oligonucleotides and
ribozymes have been used for more than a decade to target specific RNAs for degradation. Although these methods worked satisfactorily in some simple
experimental models, they have generally not delivered effective gene silencing in complex mammalian systems. However, in the past several years,
extraordinary developments in RNA interference (RNAi)-based methodologies have seen small interfering RNAs (siRNAs) become the primary means by
which most researchers attempt to target specific genes for silencing.
RNAi was first discovered in Caenorhabditis elegans, when it was noted that introducing a double-stranded RNA (dsRNA) that was homologous to a
specific gene resulted in the post-transcriptional silencing of that gene. The obvious therapeutic potential of RNAi resulted in rapid elucidation of its
mechanism of action and it is now known that gene silencing is mediated via the following basic mechanisms:
In essence, RNAi is initiated by long stretches of dsRNA that undergo processing by an enzyme referred to as Dicer. Dicer cuts the long stretches of
dsRNA into duplexes with 19 paired nucleotides and two nucleotide overhangs at both 3-ends. These duplexes are called siRNA (short interfering RNA).
The double-stranded siRNA then associates with RISC (RNA-induced silencing complex), a fairly large (approx. 160 kDa) protein complex comprising
Argonaute proteins as the catalytic core of this complex. Within RISC, the siRNA is unwound and the sense strand removed for degradation by cellular
nucleases.
The antisense strand of the siRNA directs RISC to the target mRNA sequence, where it anneals complementarily by Watson–Crick base pairing. Finally,
the target mRNA is degraded by RISC endonuclease activity.
Although early investigations found that the introduction of long dsRNA had the potential to mediate the down-regulation of any gene, it was also found
that in mammalian cells an anti-viral interferon response (IR) that resulted in the cessation of all protein synthesis was also elicited. Synthetic siRNAs
(21–23 nucleotides in length) were shown not to elicit this IR and have hence been used in studies of mammalian gene function.
This mechanism is fundamentally different from PTGS (post-transcriptional gene silencing) by ASOs (Table 1). Binding of ASOs to their target mRNA
prevents protein translation either by steric hindrance of the ribosomal machinery or induction of mRNA degradation by Rnase H (ribonuclease H).
In brief,
1. Long dsRNA precursors derived from endogenous genes or artificially introduced plasmids are cleaved by Dicer yielding siRNA.
Alternatively, synthetic siRNA can be transfected into cells.
2. siRNA is incorporated into RISC, followed by unwinding of the double-stranded molecule by the helicase activity of RISC.
3. The sense strand of siRNA is removed and the antisense strand recruits targeted mRNA, which is cleaved by RISC and subsequently degraded by cellular
nucleases.
Get the PDF File
Get the Power Point Slides