Thursday, November 28, 2019



BIOEQUIVALENCE PART 02
BIOEQUIVALENCE

PREPARED BY
MD.IMRAN NUR MANIK
LECTURER
DEPARTMENT OF PHARMACY
NORTHERN UNIVERSITY BANGLADESH
DRUG PRODUCT SELECTION
Nowadays the pharmacists are playing an important role in drug product selection. Prescription in which the product selection is permitted,
(saying that, “product selection is permitted”) pharmacist can select an equivalent drug product as he dispenses.
The hospitals or countries where there is a formulary system, the pharmacists are allowed to select an equivalent drug product from those
listed in the formulary.
Proper of a multi-source drug products is a major role and responsibility of the pharmacist. All heath practitioners should be aware that
biopharmaceutical principles can provide a sound basis for rational drug product selection.
Why important?
Drug product selection is very important because-
- The drug products from different manufacturers which contain the same amount of active ingredient may perform differently in patients.
- Differences in formulation can cause significant differences in the bioavailability of the drug. Most of the problems are associated with more compact dosage from e.g. capsules and tablets.
- Manufacturing process and materials used in the formulation of a drug product may very considerably from one drug manufacturer to other.
Therefore, it is not surprising that the various drug products of the same drug entity may exhibit different bioavailability characteristics.
Basis for drug product selection:
a. Types of cross-over study
b. Number of sample used.
c. Number of subject used.
d. Factors such as age range, weight range and sex of the subject.
e. Types of analytical procedure used.
f. The strength and type of dosage form used
B. Bioavailability data:
Blood level drug concentration and urinary drug excretion data can be used to compare the bioavailability of a specific drug form and
under various conditions. Blood level curves and urinary drug excretion profile can be used to evaluate the bioavailability of specific
drug from different drug products (various dosage forms).The data which provides the bioequivalence of drug products will be selected.
Because bioequivalent drug products maybe used as alternatives.
Data should be evaluated carefully to decide whether a drug product whether will be accepted or not.
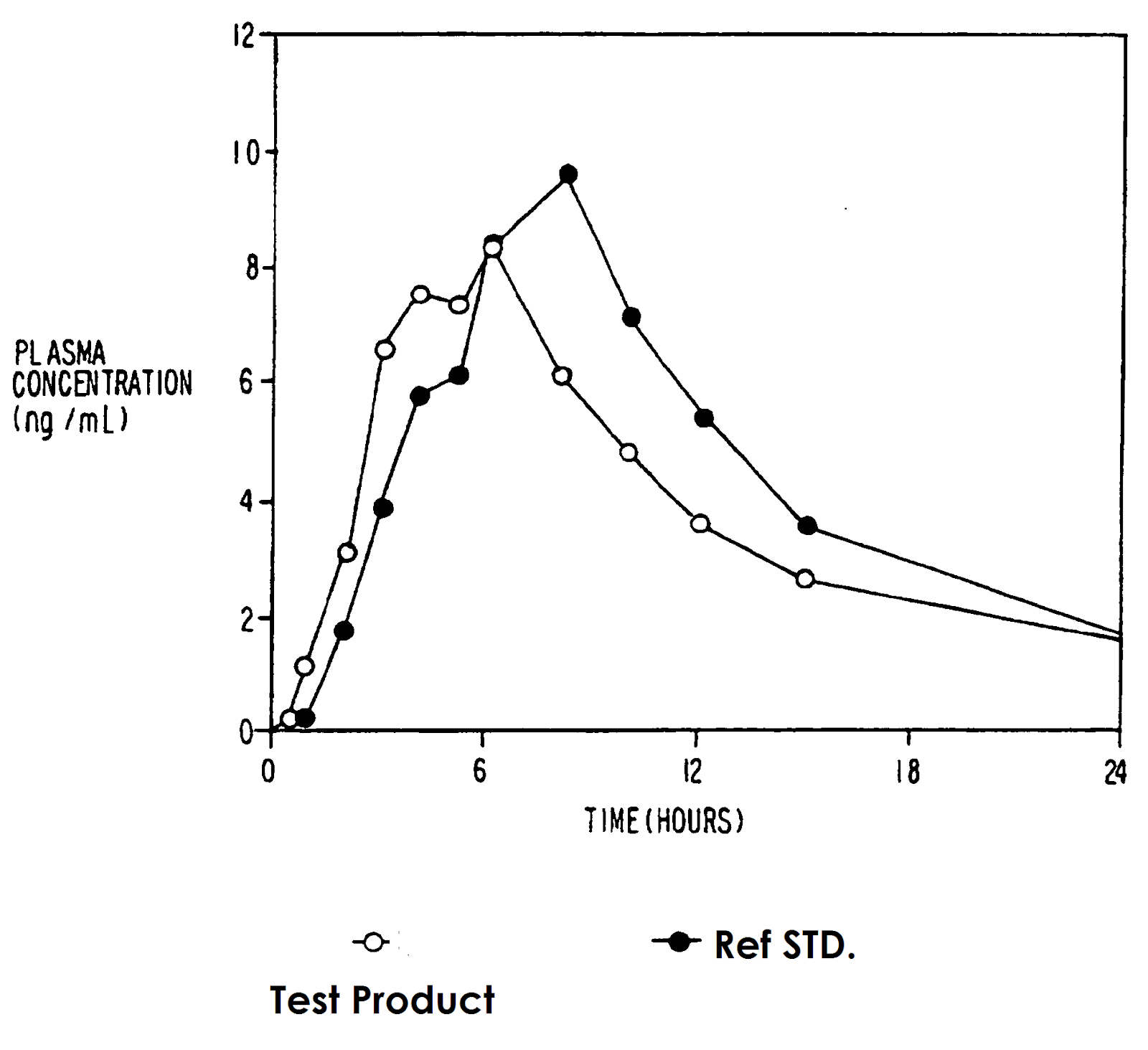
Fig. Blood level curve for meprobomate drug products.
The data in the figure indicate that the manufacturer’s drug product is equivalent to the standard reference product against which it has
been tested. The blood level of the two products is essentially super impossible. An adequate number of subjects are used in this study.
When the drug concentration time curves for two drug products are superimposable then, there is no problem to deduce that two drug
products are equivalent. Exact super-impossibility is a rare occurrence. If blood level curves for the drug products that two products of
the same active ingredient are significantly different, then it arises the question of “how much difference is allowed before one of the
products can be judged as bioequivalent to the reference standard product?”
The FDA has currently proposed a 75/75 requirement for the bioequivalence studies of certain drug products. It implies that the
relative bioavailability of test product when compare to the bioavailability of a reference products must be greater than or
equal to 75% for 75% of the subjects. When at least 75% of the subjects are administered the drug, the test drug has a bioavailability
of greater than 75% relative of the AUC and/or peak height of a reference standard.
For some drugs with a wide margin of safety e.g. penicillin, a 25% difference in the bioavailability might not significantly affect the
clinical outcome after 1-2 weeks of therapy. On the other hand, for drugs with a narrower margin of therapeutic effectiveness and
safety, e.g. digoxin a 25% decrease in bioavailability could cause significant adverse therapeutic effects.
The act of drug product selection is a legal responsibility. Professional responsibility is achieved if the practitioner selects drug
products on the basis of proper evaluation of bioequivalence data. However, care must be taken to ensure that the product has
been approved by the FDA. To properly select a drug through evaluation of bioavailability, the data is a must.
C. Other manufacturing consideration:
For many products, good bioavailability data maybe made available by many pharmaceutical manufacturers.
Therefore, other manufacturing consideration should be taken into account when selecting a manufacturer of drug products.
Some important criteria for manufacturing products are given bellows-
(1) Upon request of the pharmacist, the supplier should furnish analytical control, sterility testing and bioavailability data,
description of testing procedures for new materials and finished products or any other information which may be indicative of the
quality of a given finished drug product. This information should be supplied at no charge.
(2) The company should permit visits (during normal business hours) by the pharmacist to inspect its manufacturing and control procedures.
(3) All drug products shipped to the hospital should conform to the requirements of the most recent USP-NF criteria.
(4) The name and address of the manufacturer of the final dosage from should be present on the product labeling.
(5) Expiration dates should be clearly indicated on the package.
(After weighing all the evidence and selecting drug product, be sure to follow through on the pharmacist’s professional responsibility)
Thus, it is clear that health care practitioner must rely on their own judgment about the bioavailability of drug products they dispense.
02, Methods of determining bioavailability by clinical observation:
This method is used to establish the efficacy and safety of drugs and drug products and many such studies must be carried out by manufacturers before new product is finally approved for marketing. These studies involve a large number of patients and are very expensive. The subjects are diseased and the drug products are given to them and then asked about clinical response just before dosing, at 30 minutes and one hour after dosing and at hourly intervals, therefore a total 6 hours after dosing.
e.g. Clinical trial of analgesic zomepirac (Etodolac 400 mg) are shown in the following figure. This study used 148 points, randomly divided into five groups and each group was given one of the following drugs :- zomepirac 50 mg (Etodolac 400 mg); zomepirac 100 mg (Etodolac 200 mg) 100mg. APC with codein 60mg and placebo.
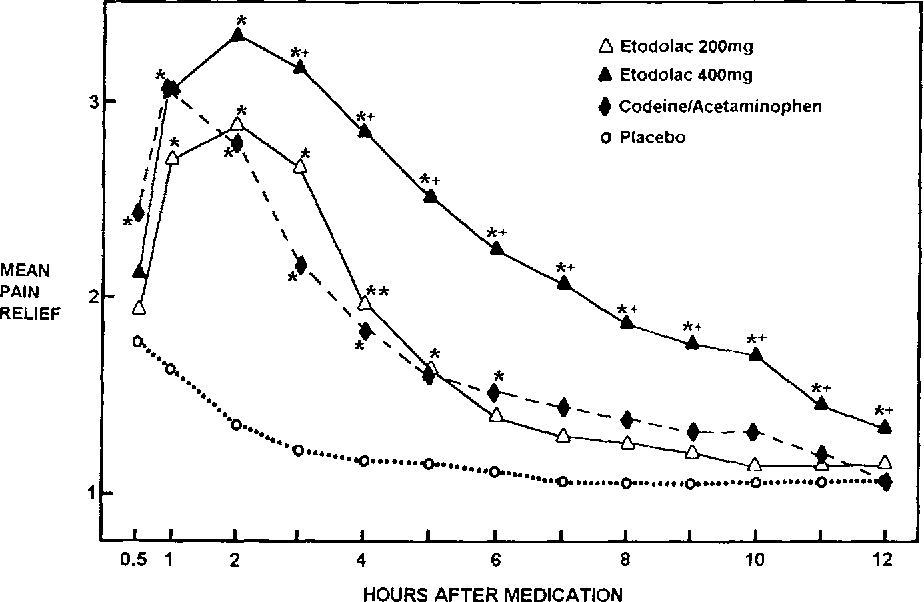
Figure I. Etodolac versus codeine/acetaminophen in the relief of pain from oral surgery (Mehlisch). *Significantly (p ~<0.05) superior to placebo. +Significantly (p ~<0.05) superior to codeine/acetaminophen. (Reproduced courtesy of the author and the editor of Rheumatology International [21])
The patients were questioned about their pain just before dosing, at 30 minutes and 1 hour after administration and at hourly intervals, therefore total of the 6 hours after dosing. At each observation patients were asked to rate their pain intensity from none to severe on a four-point scale.
The followings are the cases where bioequivalence test requirements are not relevant;
(i) Certain drug products –e.g. topically applied ointments, creams, and lotions used for local effects.
(ii) Drug products such as antacids and laxatives containing ingredient not intended for absorption.
(iii) Intravenous solution that has the same drug concentration and solvent system, as the FDA approved product.
(iv) Liquid drug products that contain the active ingredients in a solubilized form (oral solution, elixirs, syrups etc) in the same concentration as the drug product that has been the approved by the FDA via NDA ( New Drug Administration )
Types of bioavailability:-
(i) Absolute bioavailability: - When the systemic availability of a drug administered orally is determined in comparison to its intravenous administration, is called as absolute availability.
The absolute bioavailability of a drug in a drug product may be measured by comparing the respective AUC, after oral or iv administration. This measurement may be performed as long as VD & K are dependent of the route of administration.
A B A using plasma data can be determined as follows
AUC = Area under the Curve
D = Dose of administered drug
ABA using urinary drug excretion data determined by the following equation:-
The absolute bioavailability is also equal to F, the fraction of the dose that is bioavailable.
[98,99,02]Importance of bioavailability
The importance of bioavailability are given below:
1. Bioavailability is used to ensure the strength, quality, identity and purity of a drug product.
2. It evaluates the absorption, distribution, metabolism and elimination characteristics of a drug.
3. It helps to minimize the toxic effect of a potent drug .
4. It helps to select the suitable route of drug administration.
5. Dose and frequency of dose can be determined .If the bioavailability of a drug is more then the dose is less.
6. It helps to confirm the bioequivalence of drug product.
[01]Differentiate between bioavailability and bioavailable drug.
Bioavailability: The relative amount of an administered dose of a drug that reaches the systemic circulation intact and the rate at which it occurs is called bioavailability of that drug.
Thus bioavailability is concerned with the amount and rate at which the intact form of a particular drug appears in the systemic circulation following its administration.
Bioavailable dose: The fraction of an administered dose that reaches the systemic circulation intact is called bioavailable dose.
BIOEQUIVALENCE PART 01
BIOEQUIVALENCE
PREPARED BY
MD.IMRAN NUR MANIK
LECTURER
DEPARTMENT OF PHARMACY
BIOEQUIVALENCE
Define AUC with its significance.
AUC: AUC is a measurement of the extent of bioavailability of the drug. AUC is useful as a measure of total amount unaltered drug that reaches the systemic circulation. AUC is dependent on the total quantity of available drug divided by the elimination rate constant (k) and apparent volume of distribution (VD).

Where, F= Fraction of drug absorbed
Do =Total amount of drug
After IV administration F=1, because entire dose is placed into the systemic circulation. Therefore, the drug is considered to be completely available after IV administration. After oral administration of the drug F may vary from 0 to 1.
Some Definition:
(1) Bioavailability:
Bioavailability may be defined as a measurement of the rate and extent to which the therapeutically active drug reaches the systemic circulation and becomes available at the site of action from an administered dose and dosages from.
(2) Bioequivalent drug product:
Pharmaceutical equivalents or pharmaceutical alternative products that display comparable bioavailability when studied under similar experimental conditions are called bioequivalent drug products.
Bioequivalence
When two related drugs show comparable bioavailability and reach the systemic circulation at the same relative rate and extent then they are called bioequivalent and this phenomenon is called bioequivalence.
Some drugs maybe considered as bioequivalent which are equal in the extent of absorption but not in the rate of absorption. This is possible if -
- The difference in the rate of absorption is not clinically significant.
- It is not essential for the attainment of effective drug concentration in the body on chronic use.
- It is reflected in the proposed labeling.
For example, Aspirin and paracetamol are well absorbed drugs and have small differences in the rate of absorption are very little clinical consequence.
(3) Bioequivalence requirements: It is a requirement imposed by the FDA for in vitro and/or in vivo testing of specified drug products which must be satisfied as a condition for marketing.
(4) Pharmaceutical equivalents: Drug products that contain the same active ingredients (i.e. the same salt, esteron, chemical form) and are identical in strength or concentration, dosage form, and route of administration (e.g. diazepam,5mg oral tablets) are called pharmaceutical equivalents.
Chemical equivalents are pharmaceutical equivalents. pharmaceutical equivalent drug products must meet the identical standards (strength, quality, purity and identity),but may differ in such characteristics such as color, flavor, shape, scoring configuration, packaging, excipients, preservatives, expiration time and labeling.)
(5) Pharmaceutical alternatives: Drug products that contain the same therapeutic moiety but as different salts, esters or complexes are called pharmaceutical alternatives.
For example, tetracycline phosphate or tetracycline hydrochloride equivalent to 250mg tetracycline base considered pharmaceutical alternatives.
(6) Clinical equivalence: Clinical equivalence occurs when the same drug; active ingredients from two or more dosage form gives identical in vivo effects. It is measured by pharmacological responses or by controlled symptoms or disease.
(7) Pharmaceutical substitution: It is the process of dispensing a pharmaceutical alternative for (in the place of) the prescribed drug product.
For example, ampicillin suspension is dispensed in place of ampicillin capsules, or tetracycline hydrochloride is dispensed in place of tetracycline phosphate. Pharmaceutical substitution generally requires the physician's approval.
(8) Therapeutic alternatives: Drug products containing different active ingredients that are indicated for the same therapeutic or clinical objectives are called therapeutic alternatives. Active ingredients in therapeutic alternatives are from the same pharmacologic class and are expected to have the same therapeutic effect when administered to patients.
For example, cimetidine can be used in the place of ranitidine because both are H2 receptor blockers.
(9) Therapeutic equivalents: Drug products that contain the same therapeutically active ingredient that would give the same therapeutic effect and have equal adverse effects are called therapeutic equivalents. Therapeutic equivalents drug products may differ in certain characteristics such as color, scoring, flavor, configuration, and packaging, preservation and expiration date.
Therapeutic equivalent drug products must satisfy the following FDA requirements. They should be
- Safe and effective
- Pharmaceutically equivalents
- Bioequivalent
- Adequately labeled
- Manufactured in compliance with the current good manufacturing practices.
(10) Therapeutic substitution: Therapeutic substitution is the process of dispensing a therapeutic alternative in place of prescribed drug product. For example, amoxicillin is dispensed instead of ampicillin.
(11) Brand name: Brand name is the trade of the drug product. It is used by a manufacturer or distributor for identification and differentiation of the specific drug product from other competitor's products.
e.g. Napa, Nipa and APA are brand names of paracetamol and their respective companies are Beximco, Nipa and Opsonin.
(12) Chemical name: Chemical name is the name which is used by organic chemists to indicate the chemical structure of the drug/compound.
e.g. N-acetyl-para-aminophenol is the chemical name of paracetamol.
(13) Generic name: Generic name is the established nonproprietary or common name of the active drug ingredients in a drug product. e.g. acetaminophen
(14) Drug product: It is the finished dosage form that contains the active ingredient in association with additives. e.g. Paracetamol Tab, Cab. Syr.
(15) Drug product selection: The process of selecting or choosing the drug product in a specified dosage form is called drug product selection.
Relative bioavailability:
Relative bioavailability is a measure of the fraction (or percentage) of a given drug that is absorbed intact into the systemic circulation from an administered dosage form; relative to a recognized (i.e. clinically proven) standard dosage form of that drug, which is either an orally administered dosage form e.g. solution of the drug or an established suitable commercial preparation.
The relative bioavailability of two drug products given at the same dosage level and by the same route of administration can be determined with the following equation:
Where,(AUCT)test and (AUCT)standard are total areas under the plasma time concentration curve following the administration of a single dose of the test and standard dosage from respectively. This fraction may be multiplied by 100 to give percent relative availability.
When different doses of the test and standard dosage forms are administered, then a correction for the size of dose is made as follows:-
Where, D test and D standard are the sizes of the single doses of the test and standard dosage form respectively.
Using urinary data, relative bioavailability can be calculated by the following equation:-
Where [Du]α test and [Du]αStandard are the total cumulative amounts of unchanged drug ultimately excreted in urine, following the administration of single doses of the test and standard dosage form respectively.
If different doses of the test and standard dosage forms are administered, then a correction for the size of dose is made as follows:-
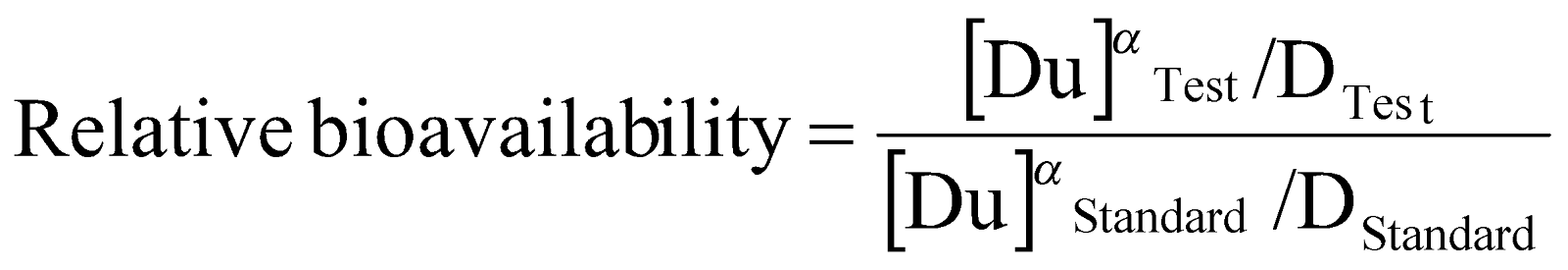
Absolute bioavailability:
The absolute bioavailability is the fraction (or percentage) of the administered dose of a given drug from a dosage form, which is absorbed intact into the systemic circulation.
Absolute bioavailability may be measured by comparing the total amount of intact drug that reaches the systemic circulation after administration of a known dose of the dosage form with the total amount of intact drug that reaches systemic circulation after administration of an equivalent dose of the drug in the form of intravenous bolus injection.
Absolute bioavailability by using plasma data can be measured as follows:-
Where, (AUCT)abs = AUC after administration of a single dose of drug via a given absorption site and (AUCT)i,v. = AUC for the drug given by via rapid intravenous injection.
If different doses of drugs are administered by both routes a correction for the sizes of the doses is made, as in the following equation:-
Using urinary data absolute bioavailability maybe calculated as follows :-

Absolute bioavailability is sometimes expressed as a percent i.e. F=1 or 100% ( F is the fraction of the dose that is bioavailable).
For drugs given intravascularly, such as by IV bolus injection, F = 1 because the entire drug is completely absorbed. For all extravascular routes of administration, such as the oral route (PO), the absolute bioavailability F may not exceed 100% (F > 1).
Practice Problem
The bioavailability of a new investigational drug was studied in 12 volunteers. Each volunteer received either a single oral tablet containing 200 mg of the drug, 5 mL of a pure aqueous solution containing 200 mg of the drug, or a single IV bolus injection containing 50 mg of the drug. Plasma samples were obtained periodically up to 48 hours after the dose and assayed for drug concentration. The average AUC values (0 to 48 hours) are given in the table below. From these data, calculate (a) the relative bioavailability of the drug from the tablet compared to the oral solution and (b) the absolute bioavailability of the drug from the tablet.
Solution
The relative bioavailability of the drug from the tablet is estimated using following equation:
No adjustment for dose is necessary.
The relative bioavailability of the drug from the tablet is 1.04, or 104%, compared to the solution.
The absolute drug bioavailability from the tablet is calculated using following equation and adjusting for the dose.
Because F, the fraction of dose absorbed from the tablet, is less than 1, the drug is not completely absorbed systemically, as a result of either poor absorption or metabolism by first-pass effect.
Methods for Assessing Bioavailability
There are several different direct and misdirect methods are available for assessing bioavailability of drug in humans. The selection of method depends on
- The purpose (objectives) of the study
- The ability of analyze the drug and metabolites in biological fluids
- The pharmacodynamics of drug substance.
- The route of drug administration
- And the nature of the drug products
The following methods are used to determine drug bioavailability of a drug from a drug product
(1) Clinical observations e.g. Well-controlled clinical trials
(2) Quantification of acute pharmacological effect.
(3) Measurement of plasma drug concentration using plasma level time curve.
4) Measurement of urinary drug excretion
(5) In vitro studies e.g. Dissolution
The following data are used when this is determined by acute pharmacodynamic effect
- Maximum pharmacodynamic effect (E max)
- Time for maximum pharmacodynamic effect
- Area under the pharmacodynamic effect time curve
- Onset time for pharmacodynamic effect
When bioavailability is determined by measuring plasma drug concentration, using plasma drug concentration time curve, the following data are used -
(a) The time for peak plasma concentration (Tmax).
(b) The peak plasma drug concentration (Cmax).
(c) The area under the plasma level time curve (AUC).
The following data are used when this is determined from urinary drug excretion
(a) The cumulative amount of drug excreted in the urine. [Du]α
(b) The rate of drug excretion in the urine (dD u/dt)
(c) The time for maximum urinary excretion (tα)
(1) Clinical observation : Well-controlled clinical trials in human is carried out to establish the safety and effectiveness of the drug product and such studies must be carried out by manufacturers before a new drug product is finally approved for marketing.
The clinical trials approach is the least accurate least sensitive and least reproductive of the general approaches for determining in vivo bioavailability. The FDA considers these approaches only when analytical & pharmacological methods are not available. Although this method would give an adequate estimation of drug in body fluids, but an adequate assay could be developed at first.
This is the most appropriates method for determining the bioavailability of topical products, where no in vivo bioavailability testing is possible.
e.g. The determination of bioequivalence of two topical antifungal products, made by different manufacturers containing the same active antifungal agent. .e.g. Ketoconazole.
Drawback: Because of high costs complexity this method is not suitable for routine bioequivalence studies.
(2) Quantification of pharmacological effect: This method assumes that a given intensity of response is associated with a particular drug concentration at the site of action. If adequate amount of drug is available at the site of action then pharmacological response is produced. The observation of more intensities of response indicates the drug is more bioavailable at the site of action. If the dose of a particular dosage form is increased the intensity of response also increases.
For example, the miotic effect after oral administration of different doses of chlorpromazine was observed and the following curve was obtained-

Drawback: This method to assess bioavailability is approved when assay methods are not available for detecting small quantity of drugs in the body fluids. Moreover, the monitoring of pharmacological data is not easy and is very difficult to establish for routine use.
There are only a limited number of pharmacological effects e.g. heart rate, blood pressure, blood sugar levels, body temperature, ECG changes that might be monitored by this method.
(3) Measurement of plasma drug concentration: Measurement of drug concentrations in blood, plasma, or serum after drug administration is the most direct and objective way to determine systemic drug bioavailability. A number of good analytical techniques such as HPLC, RIA (Radioimmunoassay), gas chromatography-mass spectroscopy (GC-MS), have been developed to detect drug and drug metabolites in blood urine.
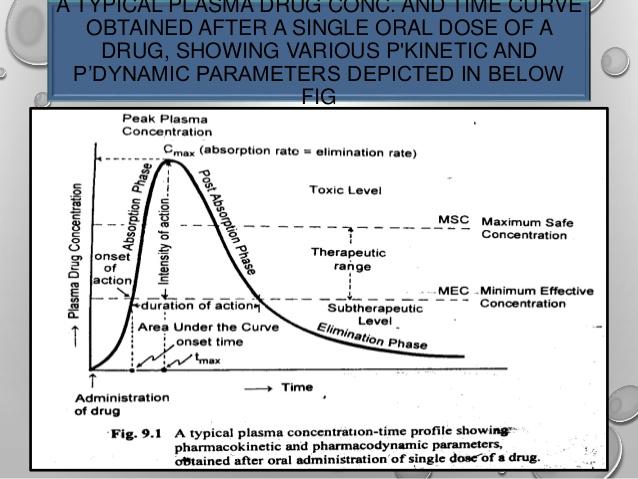
[05, 09] This is the method of choice for determining in vivo bioavailability of drug and in routine bioavailability studies because it is relatively easy to study and requires a limited number of patients; moreover it is applicable to all doses forms which are intended to deliver the drug in to the systemic circulation.
tmax :The time of peak plasma concentration.
tmax corresponds to the time required to reach maximum (peak) drug concentration in the plasma (blood) after drug administration.
Significances
- At tmax, peak (maximum) drug absorption occurs and the rate of drug absorption exactly equals the rate of drug elimination.
- tmax, indicates the rate at which a drug from a dosage form is absorbed into the systemic circulation via an absorption site.
- The drug that reaches the systemic circulation at a faster rate will have a faster onset of action.
Units for tmax are: hours and minutes.
Cmax : The peak plasma drug concentration.
Cmax, represents the maximum plasma drug concentration obtained after oral administration of drug.
Significances
- For many drugs, a relationship is found between the pharmacodynamic drug effect and the plasma drug concentration.
- Cmax provides indications that the drug is sufficiently absorbed systemically to provide a therapeutic response.
- In addition, Cmax provides warning of possibly toxic levels of drug.
Units for Cmax are: concentration units (e.g. mg/mL, ng/mL).
AUC: The area under the plasma level time curve.
AUC is a measurement of the extent of drug bioavailability.
Significances
- The AUC reflects the total amount of active drug that reaches the systemic circulation.
- The AUC is the area under the drug plasma level time curve from t = 0 to t =α, and is equal to the amount of unchanged drug reaching the general circulation divided by the clearance.
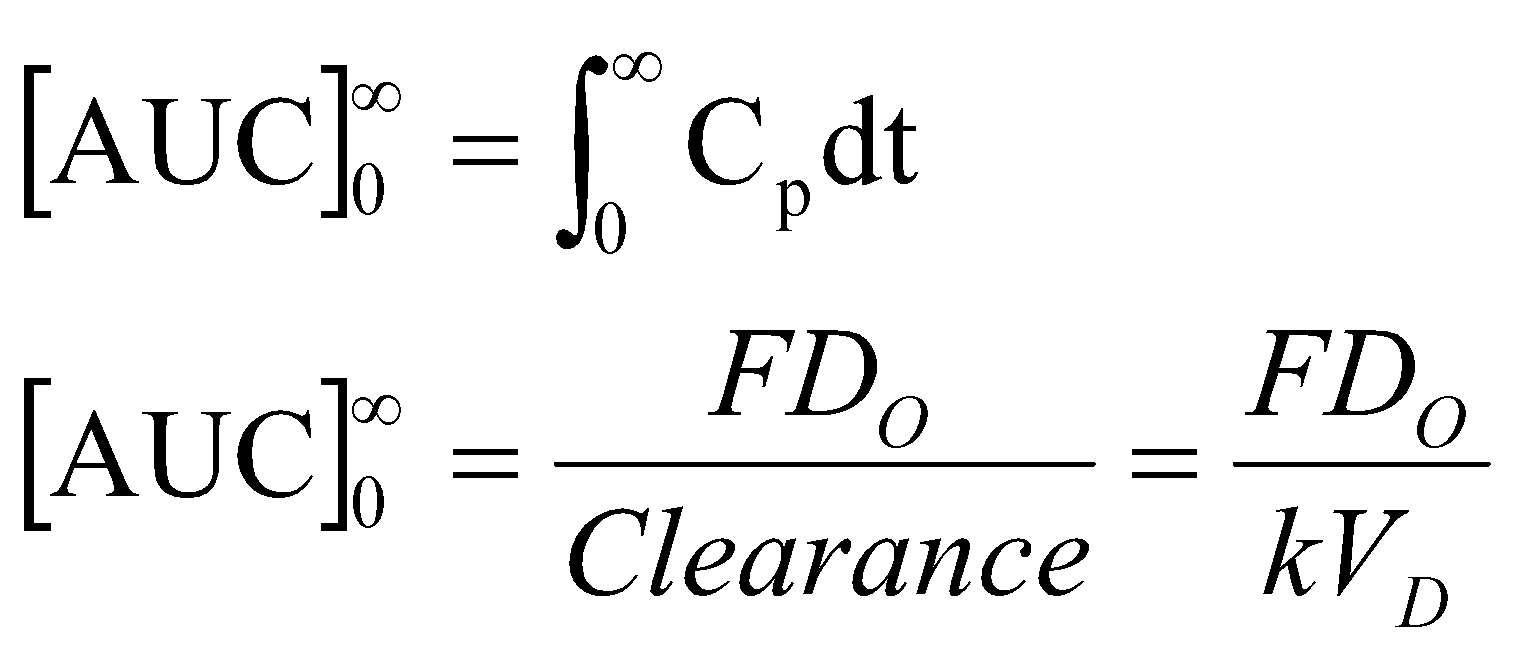
Where F = fraction of dose absorbed, D0 = dose, k = elimination rate constant, and VD = volume of Distribution.
The AUC can be determined by a numerical integration procedure, such as the trapezoidal rule method. Units for AUC are: concentration time (e.g. μg hr/mL).
iii) AUC is independent of route of drug administration and drug elimination process but dependent on dose of administration until the the elimination process unchanged. For example, if a single dose of a drug is increased from 250 to 1000 mg, the AUC will also show a fourfold increase.
iv) In some cases, the AUC is not directly proportional to the administered dose for all dosage levels. For example, as the dosage of drug is increased, one of the pathways for drug elimination may become saturated, then AUC does not increases proportionally with the dose , because a small amount of drug is being eliminated and more drug is retained.
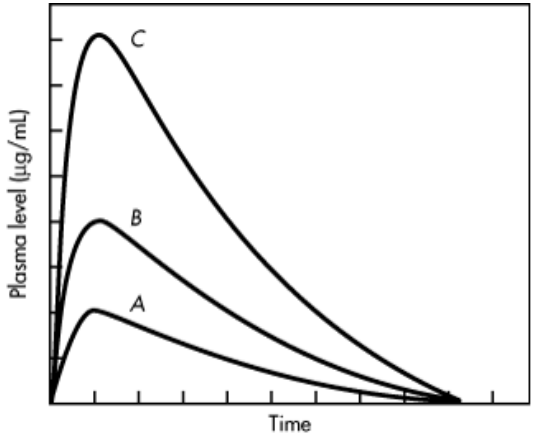
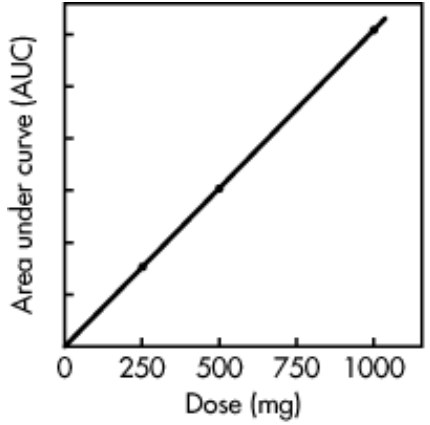
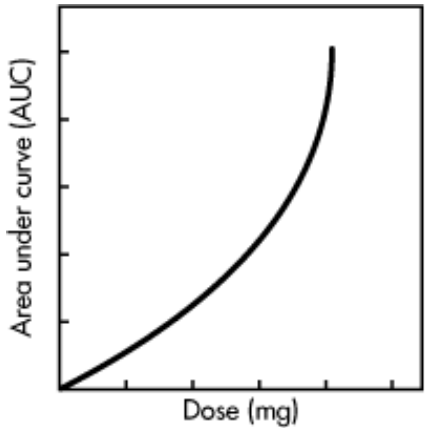
Fig A: Plasma level time curve following administration of single doses of (A) 250 mg, (B) 500 mg, and (C) 1000 mg of drug.
Fig B: Linear relationship between AUC and dose (data from).
Fig C: Relationship between AUC and dose when metabolism is saturable.
Drug elimination includes the processes of metabolism and excretion, while drug metabolism is an enzyme-dependent process. For drugs such as salicylate and phenytoin, continued increase of the dose causes saturation of one of the enzyme pathways for drug metabolism and consequent prolongation of the elimination half-life. Thus the AUC increases disproportionally with the increase in dose, because a smaller amount of drug is being eliminated (i.e., more drug is retained).
Disadvantages: When the AUC is not directly proportional to the dose, bioavailability of the drug is difficult to evaluate because drug kinetics may be dose dependent.
(4) Measurement of bioavailability by Urinary Drug Excretion Data: Determination of bioavailability by urinary drug excretion data is an indirect method. For estimating bioavailability the drug must have the following characteristics;
- The drug must be excreted in significant quantities as unchanged drug in the urine.
- Urine samples must be collected timely. and
- The total amount of urinary drug excretion must be obtained.
In this case the following data are used.
Duα: The cumulative amount of drug excreted in the urine.
Duα is directly related to the total amount of drug absorbed. That is the higher the cumulative value, the higher the drug absorbed.
Procedure to obtain Duα : Experimentally, urine samples are collected periodically after administration of a drug product. Each urine specimen is analyzed for free drug using a specific assay method. Then the cumulative amount of drug excreted in the urine is plotted against time, fig.B.
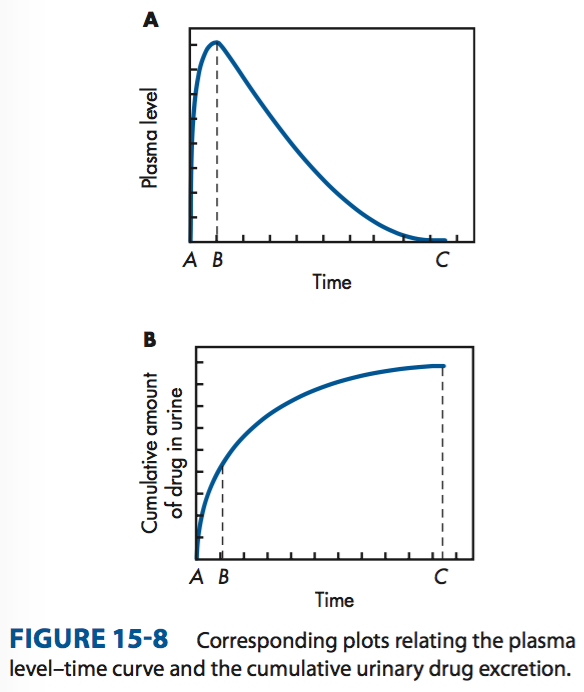

Suppose a single dose of two tablet formulation (Tablet A and Tablet B) were administered and when the urine sample was analyzed then fig.C was obtained. The curves in fig. C shows that the drug in a tablet A has a much higher cumulative value then drug in tablet B. Hence, the drug in tablet A is much more absorbed then from tablet B.
When the drug is completely eliminated pint C in the fig. A& B, the plasma concentration approaches 0 and the maximum amount of drug excreted in urine (Duα ) is obtained.
dDuα/dt: The rate of drug excretion in the urine
As most of the drugs are eliminated by a first-order rate process, the rate of drug excretion (dDuα/dt) is dependent on the first-order elimination rate constant k and the concentration of drug in the plasma Cp.
So, when dDuα/dt (rate of drug excreted in urine) reaches its maximum level, it indicates the Cmax level. Because at Cmax the drug reaches its maximum level in plasma, so at this level the rate of drug excretion would be maximum according to first-order elimination.
In the figure fig. A& B the maximum rate of drug excretion at point is at point B, whereas the minimum rate of drug excretion is at points A and C. Thus, a graph comparing the rate of drug excretion with respect to time should be similar in shape as the plasma level time curve for that drug.
t∞ The total time required for the drug to be excreted.
In the above figures (fig. A& B ) the slope of the curve segment from A to B is related to
the rate of drug absorption, whereas point C is related to the total time required (after drug administration) for the drug to be absorbed and completely excreted ( t = ∞) . The t∞ is a useful parameter in bioequivalence studies that compare several drug products.
the rate of drug absorption, whereas point C is related to the total time required (after drug administration) for the drug to be absorbed and completely excreted ( t = ∞) . The t∞ is a useful parameter in bioequivalence studies that compare several drug products.
According to first order kinetics the drug will not completely eliminated, so the “t” is never zero and will not touch the X axis.
Methods and criteria for bioavailability testing:
[09] Method: The general method of bioavailability testing involves:
(i) Administering a drug to a healthy human subject.
(ii) Obtaining serial blood or urine samples over a period of time.
(iii) Analyzing the samples for drug content.
(iv) Tabulating and graphing the results.
(v) Statistical tests are used to determine if any differences observed during the study.
[08] Criteria: The criteria’s of bioavailability testing are:
(i) Test should be conducted in a cross-over design to minimize the effect of individual subject variation to drug.
(ii) At least 12 subjects should be used, although 18 to 24 subjects are used to increase the data base for statistical analysis.
(iii) Informed, written consent should be taken from each of the individual/subject.
(iv) Adequate examination and laboratory tests (hematology, blood chemistry and urine analysis) are carried out to establish, the human subjects as healthy volunteers.
(v) Healthy human subjects should weigh between 55-95 kg and the individual weight of subjects should be closed to the desirable weight for height frame and age.
(vi) The subjects should not take any drugs prior and during the study.
(vii) Food and fluid intake prior to and during the test must be kept uniform.
(viii) Usually the volunteers fast overnight and should take the drug at first in the morning with prescribed amount of water.
(ix) A time period of 3 weeks equivalent to 10 average half-lifes, should separate the cross over test.
(x) Test should be conducted compared with the reference standard products.
(xi) Test should be conducted in a suitable time period so that adequate data such as serum drug concentration vs time and urine drug concentration vs time can be obtained; from which peak serum drug level Cmax and its time, tmax and AUC can be obtained.
(xii) For the following information; different studies are used to determined bioavailability:-
(a) The drug product for topical preparation.
(b) The drug product which are administered orally but is not intended to be absorbed (antacid).
(c) The drug products which are administered by inhalation as a gas or vapor (e.g. inhaler).
Preparation of bioequivalence data: Bioequivalence study data should be presented so that the important factors like -
- Peak serum concentration (Cmax),
- The time for peak plasma concentration (tmax),
- AUC : Area Under Plasma Drug Concentration curve can readily and adequately be evaluated.
This presentation usually involves a format that includes- (i) tabulated data and (ii) drug concentrations versus time figure.
The following factors are included to the formula representing bioequivalence data -
1. Name and manufacturer of drug
2. Dosage form of drug.
3. Number of volunteers and number of each sex.
4. Types of analytical producers used in the study.
5. Type of study.
6. Sampling intervals.
7. Drug concentration at each sampling intervals.
8. Average of individual peak serum drug concentration.
9. Average of the times for peak serum drug concentration.
10. AUC for the various time intervals. (Ave. AUC)
11. Statistical method used to evaluate the data.
12. The result of the statistical evaluation.
Cross-over design: A simple study design can lead to false information due to 3 reasons as follows:-
(i) Different subject population,
(ii) Different study conditions,
(iii) Different assay methodology.
The errors arising from the above three causes may overcome by studying cross-over design.
In the cross-over design, all individual subjects receive each formulation once. A suitable cross-over design should be utilized in bioavailability testing, so that subjects daily variation are equally distributed among all dosage forms or drug products being tested.
Types of cross-over designs: There are three types of cross-over designs;-
Types of cross-over designs: There are three types of cross-over designs;-
1. Four-way cross-over design.
2. Three-way cross-over design
3. Two-way cross-over design.
1. Four-way cross-over designs: In a 4-way cross-over design, four different drug foundation
(A, B, C, D), are used in 12 volunteers , divided in 4 groups for the bioavailability study. This type of design plans the clinical trail so that each subject receives each drug product only once. An adequate time is allowed between the medications (two successive administration of the drug) for the elimination of the drug from the body.
(A, B, C, D), are used in 12 volunteers , divided in 4 groups for the bioavailability study. This type of design plans the clinical trail so that each subject receives each drug product only once. An adequate time is allowed between the medications (two successive administration of the drug) for the elimination of the drug from the body.
Thus the drug product B may be followed by drug product A, D or C. A four way cross over design may be described as follows:-
Table : 4-way cross-over design for a bioequivalence study of 4 different drug products in 12 human volunteers dividing them into 4 groups. All subject (each group) received each drug product only once without repetition.
After each subject receives a drug product, the blood samples are collected at an appropriate time intervals so that a blood drug level-time curve is obtained. The time intervals should be sufficiently spaced so that the peak blood concentration, the AUC, and the absorption and elimination phases of the curve may be well described. In some cases, the measurement of drug in urine samples may be necessary.
(ii) Three-way crossover design:
In a three-way cross-over design, three different drug foundation (say A,B,C) are used in 6 volunteers for a bioequivalence study. In this design, each subject receives each drug product only once, with adequate time between medications for the elimination of the drug from the body. Thus, drug product a may be followed by B or C.
A three way cross-over design maybe expressed as the following tabulating form:-
Table : A three-way cross-over design for a bioequivalence study of 3 drug products in 6 human volunteers.
(iii) Two-way crossover design:
In a two-way crossover design, two different drug products 9A,B) are used in two groups of volunteers or two volunteers.
Table: A two-way cross-over design for a bioequivalence study of 2 different drug products.
Importance of crossover designs:
(i) These designs reduce subject to subject variation in bioequivalence study.
(ii) Variation due to sequence, period, and treatment (formulation) is reduced; because all patients do not receive the same drug product on the same day and in the same order
(iii) Possible carry-over effects from any particular drug product are minimized by changing the sequence or order in which the drug products given to the subject.
Advantage:
The limitation of simple bioavailability study can be overcome by using this process.
[02] Wash-out period:
The time during which most of the drugs are eliminated from the body is called the washout periods. Generally about it is about 10 elimination half-lives of a drug product.
[02] Question: Theophylline has a half-life of 3 hours. Calculate the wash out period of theophylline.
Answer: We know, wash out period is about 10 elimination half life of a drug product.
Thus the wash out period of theophylline= 10 t1/2=10×3=30 hours.
[01,04,07,09] Purposes of bioavailability studies :
(i) Bioavailability studies are performed for both approved active drug ingredients and therapeutic moieties which are yet to be approved for marketing by FDA. To be approved by the FDA, the drug products must be safe and effective and must meet all applicable standards of strength, quality and purity. To ensure these standards the FDA requires bioavailability or pharmacokinetic studies and where necessary bioequivalence studies for all drug products.
(ii) For un-marketed drug products, in-vivo and in-vitro bioequivalence studies must be performed on the drug formulation proposed for marketing as a generic drug product. Furthermore, the essential pharmacokinetic parameters of the active drug ingredient or therapeutic moiety must be characterized, Essential pharmacokinetic parameters including the
✓Rate and extent of systemic drug absorption,
✓Rates of excretion & metabolism and
✓Elimination half-life should be determined after administration of single as well as multiple doses. The bioavailability study data are important to establish recommended dosage regimens and to support drug labeling.
(iii) In-vivo bioavailability studies are also performed for new formulations or therapeutic moieties that are fully approved for marketing. The purpose of these studies is to determine the bioavailability and to characterize the pharmacokinetics of the new formulations, new dosage from, new salt or ester with respect to a reference standard formulation.
(iv) Clinical bioavailability studies are useful in determining the safety and efficacy of the drug products.
(v) Bioavailability studies are used to define the effect of changes in the dosage form and physicochemical properties of drug products on the pharmacokinetic of the drug.
(vi) Bioequivalence studies are used to compare the bioavailability of the same drug (same salt or ester) from various drug products.
(vii) Bioavailability and Bioequivalence can also be considered as performance measures of the drug products in-vivo.
Answer: [2000, 99] Bioequivalence requirement: A requirement imposed by the FDA for in-vitro and/ or in-vivo testing of specified drug product. This requirement must be satisfied as a condition for marketing.
[2000, 97] Criteria for establishing a bioequivalence requirement: Bioequivalence requirements may be imposed by FDA on the basis of the following:
1. Evidence from well controlled clinical trials, or controlled observations in patients and bioequivalence studies:
- Various drug products do not give comparable therapeutic effects are not bioequivalent drug products.
- Narrow therapeutic ratio and minimum effective concentration in the blood.
- Serious adverse effects.
2. Physicochemical:
- Low solubility in water.
- Dissolution rate slow.
- Particle size and surface area of the effective drug ingredient.
- Structural forms dissolve slowly.
- High ratio of excipients.
- Hydrophilic or hydrophobic excipients and lubricants
3. Pharmacokinetic:
- GI or localized site
- Degree of absorption poor.
Period: Period refers to the time period in e a study is performed. A two period study is a study that is performed on 2 different ways.
Sequence: A sequence refers to the no of different orders in the treatment groups in a study.
For groups a two sequence, two period studies would be designed as follows:-
Where, R = Reference and T= Treatment
Subscribe to:
Posts (Atom)