Cardiovascular Drugs
(Medicinal Chemistry)

Cardiovascular Drugs
(Medicinal Chemistry)
Blood pressure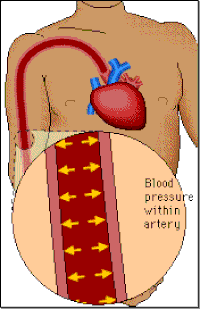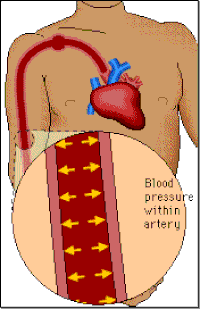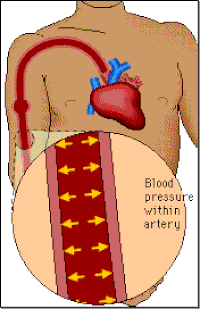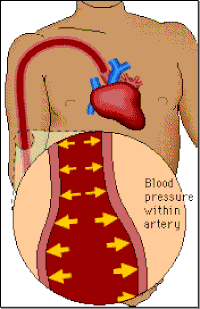
Blood pressure is a measure of the force of the blood pushing against the walls of the arteries (i.e. the blood vessels that carry blood from the heart to the rest of the body). It is determined by the amount of blood pumped by the heart, the pumping power of the heart, the condition of the heart valves and the size and condition of the arteries.
Systolic Blood Pressure (SBP):
When the heart contracts to pump out blood, pressure is highest. This measurement is called the systolic pressure.
Diastolic Blood Pressure (DBP):
After pumping, the heart relaxes and pressure drops to its lowest point just before new beat starts. This measurement is called the diastolic pressure.
Hypertension
Hypertension or High Blood Pressure is the name of a pathological condition in which blood pressure is persistently elevated (i.e. it stays high for a long period of time).
Hypertension is the most common cardiovascular disease. Persistent rise of blood pressure above normal upper level (120/80 mm Hg) according to age and sex is known as hypertension.
Definition
Hypertension is defined as either a sustained systolic blood pressure (SBP) of greater than 140 mm Hg or a sustained diastolic blood pressure (DBP) of greater than 90 mm Hg.
Classification of HP
According to WHO, the systolic and diastolic blood pressure in normal adult is equal to or below 140 mm Hg and 90 mm Hg. The blood pressure may be of three types.
- Mild hypertension - 90-104mmHg
- Moderate hypertension 105-114mmHg
- Severe hypertension above 115mmHg
Secondary hypertension
A specific cause of hypertension can be established in only 10-15% patients. Patients in whom specific cause of hypertension can be found are said to have secondary hypertension.
Essential, idiopathic, or primary hypertension
On the other hand 85 to 90% patients in whom no specific cause of hypertension can be found are said to have essential or primary hypertension.
Antihypertensive agents
Drugs which lower the elevated blood pressure to normal level and which are used for the treatment of hypertension are called antihypertensive drugs.
B.P = Cardiac output (CO) ☓Peripheral resistance
|
The drugs act by reducing cardiac output or reducing the total peripheral resistance without correcting the cause of hypertension.
Figure : Major factors influencing blood pressure
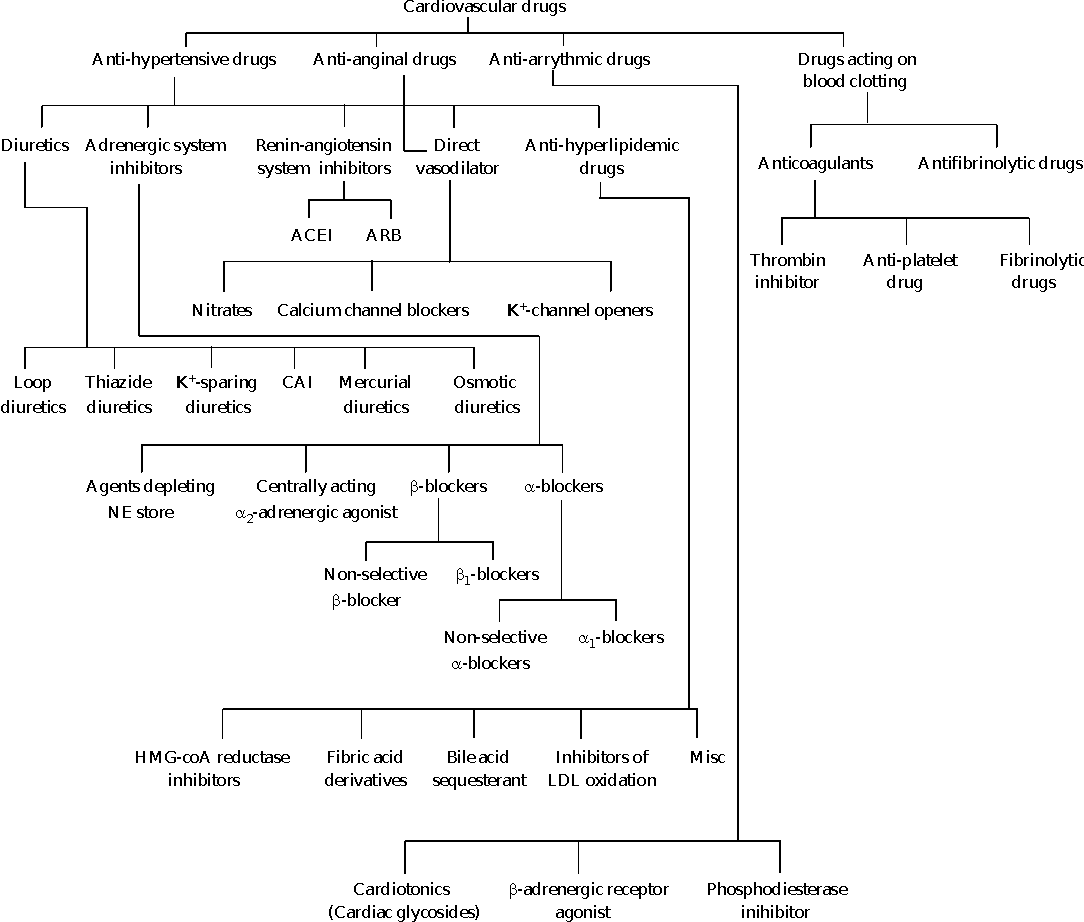
Classification of anti-hypertensive agents
- Adrenoreceptor blocking agents:
Sympatholytic agents: Enhances BP, lowers reflexes, Stimulates heart rate.
- Centrally acting sympatholytic agents.
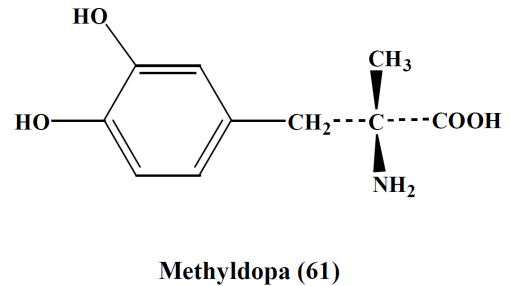
- Methyldopa,
- Clonidine

- β – adrenergic receptor antagonists (β – blockers)
- Propranolol, labetalol (non selective β –blockers)
- Metoprolol, atenolol (β1 –selective)

Figure: β-adrenergic receptor antagonists (β-blockers)
- α– adrenergic receptor antagonists (ARB)
- Prazosin
- Terazosin
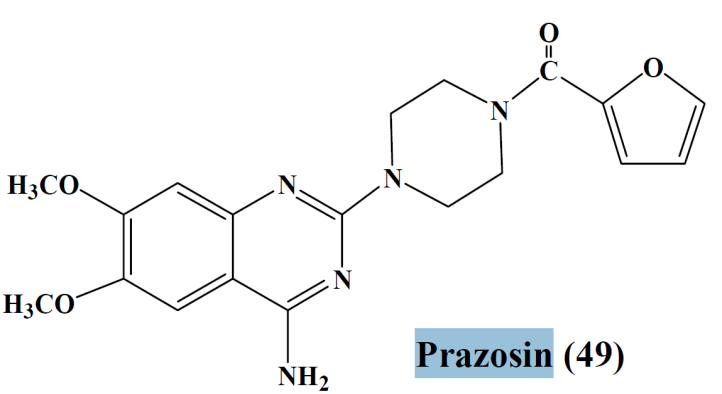
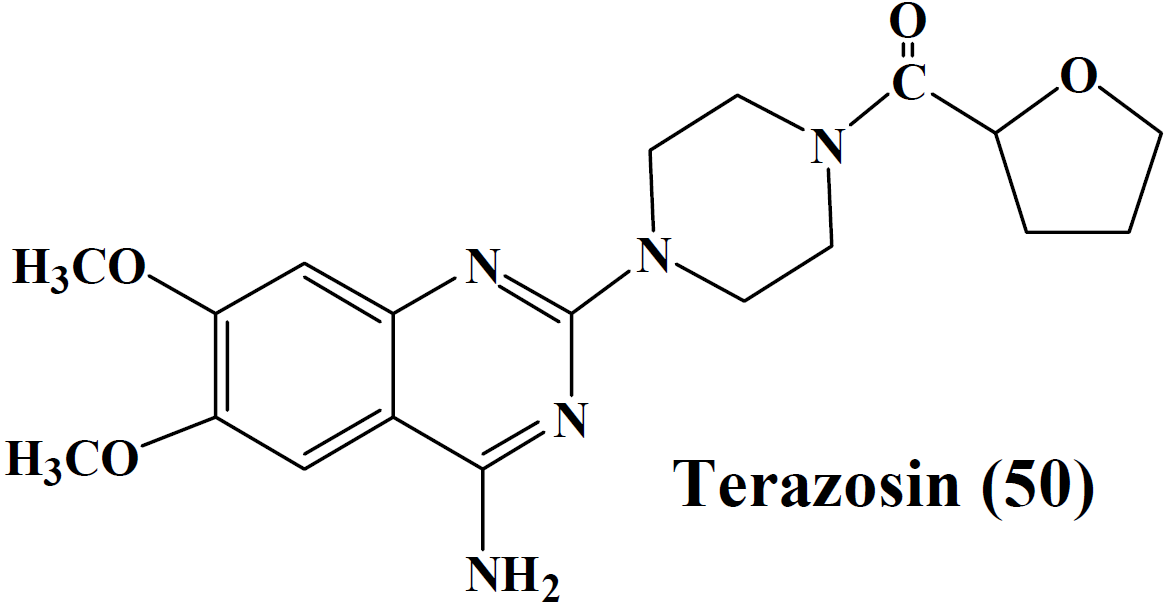
Fig:Sodium nitroprusside
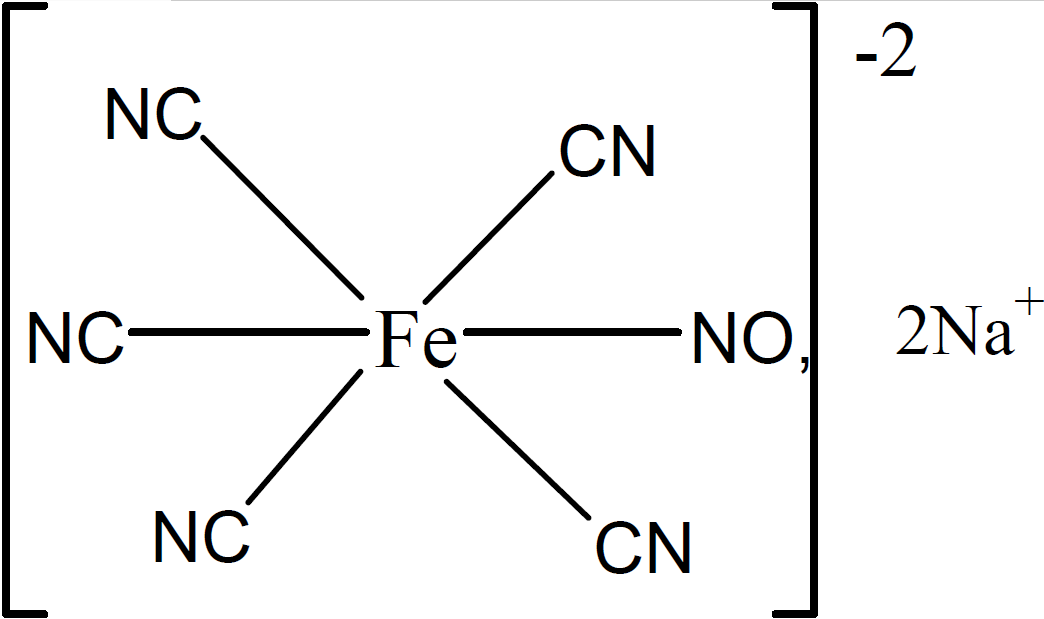 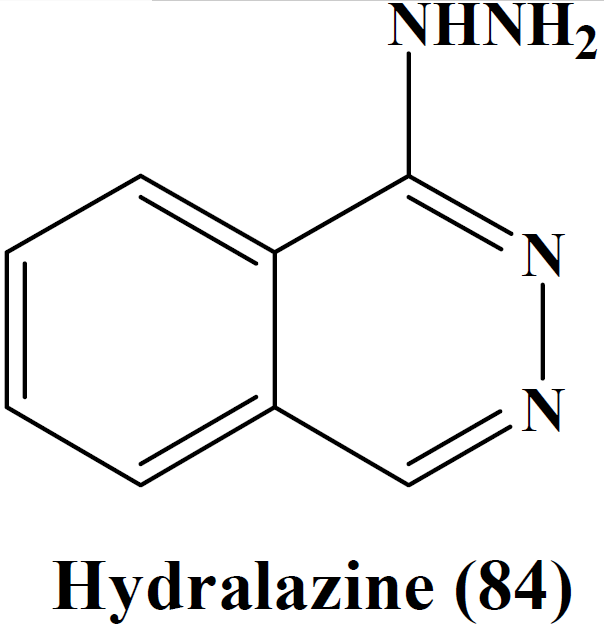 |
2) Vasodilators
- Direct vasodilators
- Hydralazine,
- Sodium nitroprusside
- K+ channel antagonist
- Minoxidil
- Diazoxide

Fig: Diazoxide Fig: Minoxidil
- Ca++ channel blockers
- Nifedipine,
- Verapamil,
- Diltiazem
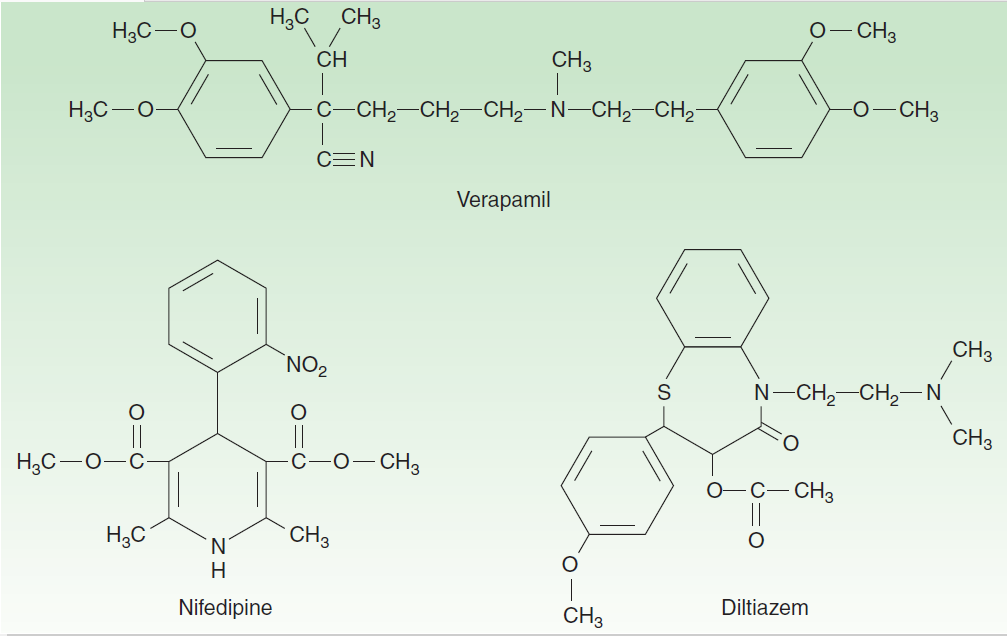
Figure: Ca++ channel blockers
3) Agents acting on rennin-angiotensin system:
A. Angiotensin converting enzyme inhibitors (ACE inhibitor)
- Captopril,
- Enalapril
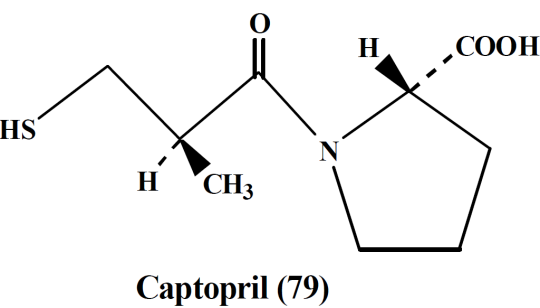
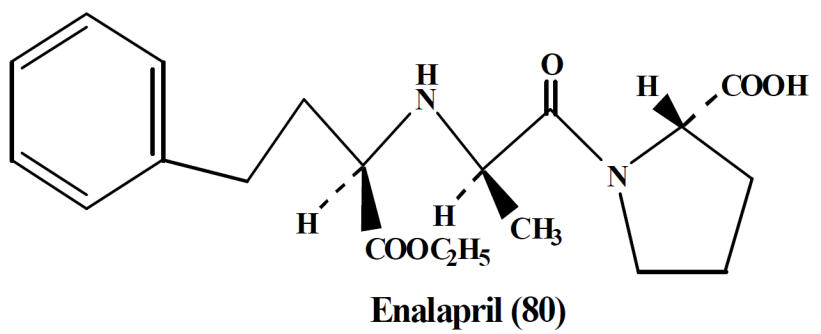
B. Angiotensin II inhibitors
- Losartan

- Diuretics:
a) Thiazides: e.g Hydrochlorothiazide
b) Loop Diuretics: e.g. Furosemide
c) Potassium Sparing Diuretics: e.g. Triamterene, Spironolactone
•Structures to be concluded from the diuretic chapter.
☠ Do Not Forget the Diuretics ☠
β –blockers
β–receptor blockers are the drugs that selectively and competitively block the actions of catecholamines mediated through β – receptor stimulation.
Classification of β – adrenergic blocking agents:
- Nonselective β–(β1 + β2) adrenergic antagonists:
- Propranolol
- Labetalol
- Nadolol
- Timolol
- Pindolol
- Selective β1–adrenergic antagonists:
- Atenolol
- Metoprolol
- Esmolol
- Acebutolol
Effect of β adrenergic receptor blockers
The β-blockers reduce blood pressure primarily by decreasing cardiac output (Figure). They may also decrease sympathetic outflow from the central nervous system (CNS) and inhibit the release of renin from the kidneys, thus decreasing the formation of angiotensin II and the secretion of aldosterone. The prototype β-blocker is propranolol
[proe PRAN-oh-lol], which acts at both β1 and β2 receptors.
[proe PRAN-oh-lol], which acts at both β1 and β2 receptors.
Selective blockers of β1 receptors, such as metoprolol [met-OH-pro-lol] and atenolol
[ah-TEN-oh-lol], are among the most commonly prescribed β-blockers.
The selective β-blockers may be administered cautiously to hypertensive patients who also have asthma, for which propranolol is contraindicated due to its blockade of
β2-mediated bronchodilation.
[ah-TEN-oh-lol], are among the most commonly prescribed β-blockers.
The selective β-blockers may be administered cautiously to hypertensive patients who also have asthma, for which propranolol is contraindicated due to its blockade of
β2-mediated bronchodilation.
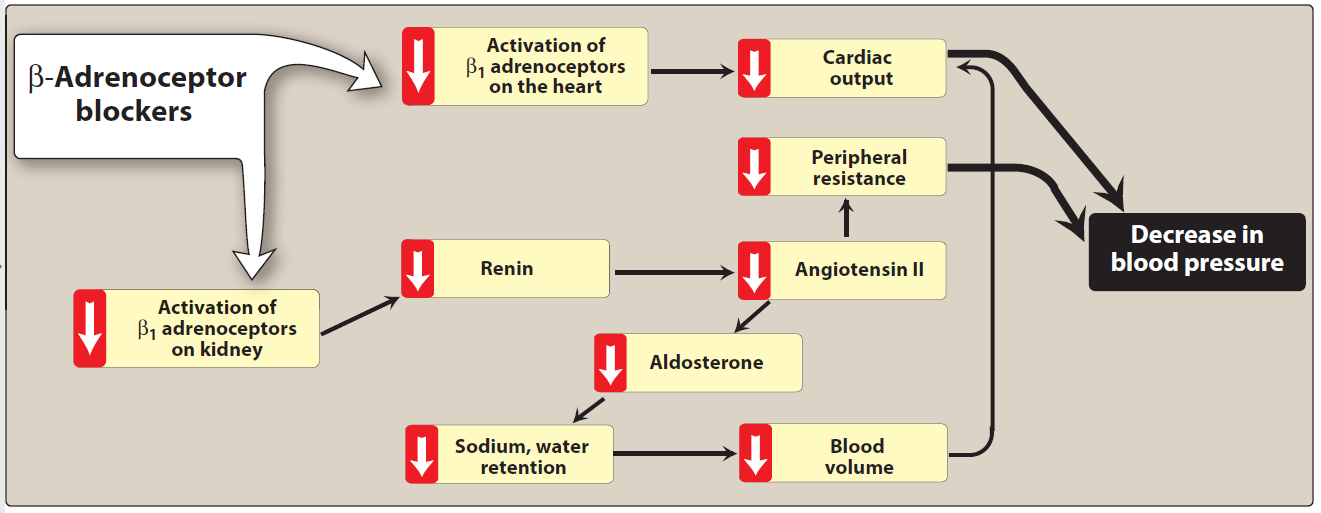
Figure: Actions of β-adrenoceptor–blocking agents.
Propranolol
Propranolol was the first β –blocker shown to be effective in hypertension.
Propranolol is a competitive β –adrenergic antagonist. It has equal affinity for β1 – and β2 –receptors; thus, it is a nonselective β – adrenergic receptor antagonist. Propranolol decreases blood pressure primarily as a result of a decrease in cardiac output. The major therapeutic effect of propranolol is on the cardiovascular system. β1 –receptor is present in myocardium. Propranolol blocks the β1 – receptor in heart. Since catecholamines have positive chronotropic and inotropic actions, β – adrenergic antagonist propranolol slows the heart rate and decreases myocardial contractility, decreases cardiac output & decreases blood pressure.
Nonselective β –adrenergic antagonists such as propranolol block β2 – adrenergic receptors in bronchial smooth muscle. This usually has little effect on pulmonary function in normal individuals. However, in patients with asthma or chronic obstructive pulmonary disease, such blockade can lead to life-threatening broncho-constriction.
Metoprolol

Metoprolol is a β1 – selective adrenergic antagonist.
It is approximately equipotent to propranolol in inhibiting stimulation of β1 –adrenoceptors such as those in the heart but 50 to 100 fold less potent than propranolol in blocking β2 – receptors. Although metoprolol is in other aspects very similar to propranolol, its relative cardioselectivity may be advantageous in treating hypertensive patients who also suffers from asthma, diabetes, or peripheral vascular disease.
It is approximately equipotent to propranolol in inhibiting stimulation of β1 –adrenoceptors such as those in the heart but 50 to 100 fold less potent than propranolol in blocking β2 – receptors. Although metoprolol is in other aspects very similar to propranolol, its relative cardioselectivity may be advantageous in treating hypertensive patients who also suffers from asthma, diabetes, or peripheral vascular disease.
Studies of small number of asthmatic patients have shown that metoprolol causes less bronchial constriction than propranolol at doses that produce equal inhibition of β1 – adrenocepotor responses.
Cardioselective β – antagonists:
Cardioselective β – antagonists are drugs that have a greater affinity for the
β1 – receptors of the heart than for β2 – receptors in other tissues, such cardioselective agents should provide two important therapeutic advantages.
β1 – receptors of the heart than for β2 – receptors in other tissues, such cardioselective agents should provide two important therapeutic advantages.
- The first advantage would be the lack of an antagonistic effect on the
β2 – receptors in the bronchi. Theoretically, this would make β1 – blockers safe for use in patients who have bronchitis or bronchial asthma. - The second advantage would be the absence of blockade of the vascular
β2 – receptors, which mediate vasodilatation. This would be expected to reduce or eliminate the increase in peripheral resistance that sometimes occurs after the administration of nonselective β – antagonists.
Ca+2 channel blockers
Classification: Chemical classification
A) Phenylalkylamines : e.g. Verapamil
B) 1,4-dihydropyridines : 1st Gen → Nifedipine
2nd Gen → Isradipine, Nicardipine, Felodipine
3rd Gen → Amlodipine
C) Benzothiazepines: e.g. Diltiazem
D) Newer second generation alkyl amine derivative: e.g. Bepridil
Second generation is more potent analogues than first generation

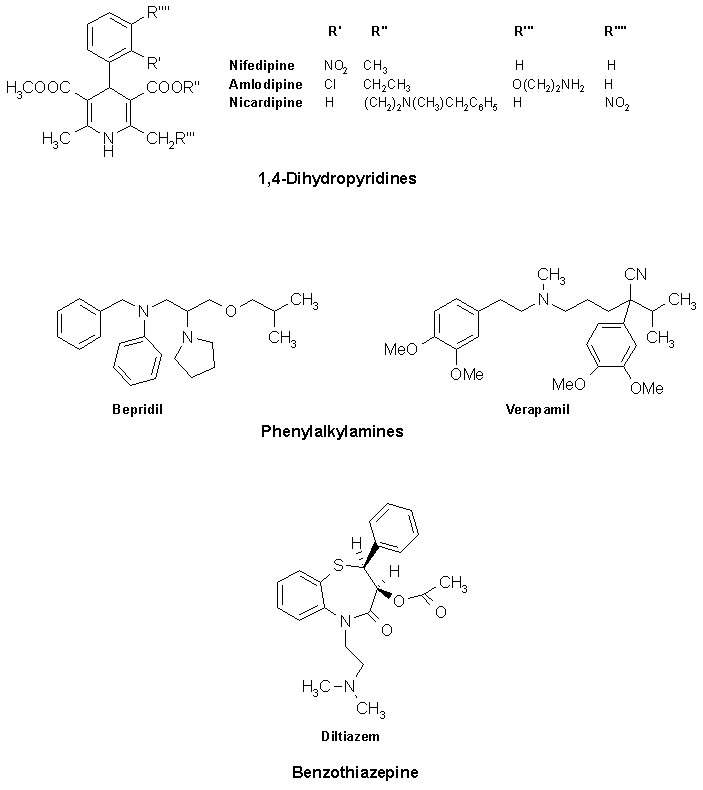
Chemistry
Diltiazem and verapamil are both chiral, possessing asymmetric centers. In each case, the dextro-rotatory
(i.e. the (+) enantiomer) is approximately 10 times more potent as a calcium channel blocker than the levo-rotatory (i.e. (–)-enantiomer).
(i.e. the (+) enantiomer) is approximately 10 times more potent as a calcium channel blocker than the levo-rotatory (i.e. (–)-enantiomer).
Amlodipine is used therapeutically as a racemic mixture, composed of S- and R-enantiomers, but its calcium channel-blocking effect is confined to S-amlodipine; R-amlodipine has 1000-fold less activity than its S-enantiomer. The affinity of its levorotary (-)-enantiomer to the calcium channels is 1,000 times superior to that of its dextrorotary (+)-enantiomer.
Role of calcium in muscle contraction
Calcium channels
A calcium channel is an ion channel which displays selective permeability to calcium ions. It is sometimes synonymous as voltage-dependent calcium channel although there are also ligand-gated calcium channels.
Types: There are 5 types of calcium channel
- L type: located in skeletal, cardiac and smooch muscles
- T type: found in pacemaker cells
- N type: found in neurons
- P type: located in purkinje cells
- R-type : Cerebellar granule cells
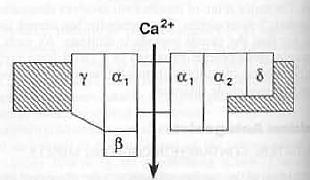
Among the Ca-channels L-type calcium channel is the target for calcium channel blockers which consists of 5 peptide subunit: α1, α2, β, γ, δ. α1 provides central pore of the channel while other subunits cover α1 and maintain lipophilicity of the cell membrane. Fig: L-type channel
Mechanism of action
- Calcium enters cells by voltage-sensitive channels and by receptor-operated channels that are controlled by the binding of agonists, such as catecholamines, to membrane receptors. The calcium channel blockers inhibit vascular
smooth muscle contraction by depriving the cell from the calcium ions.
- Calcium-channel blockers, such as verapamil and diltiazem, are more effective against the voltage-sensitive channels, causing a decrease in the slow inward current that triggers cardiac contraction. Verapamil and diltiazem are ionic at physiological pH so that they enter into the binding site when channel is open.
- Nifedipine (Dihydropyridines) is neutral at physiological pH. It interacts with
Ca channel when it is open or closed due to its lipophillic nature.
- Verapamil and diltiazem by binding to the open, depolarized channels, prevents repolarization until the drug dissociates from the channel. By decreasing the inward current carried by calcium, verapamil and diltiazem slow conduction and prolong the effective refractory period in tissues that are dependent on calcium currents, such as the AV node.
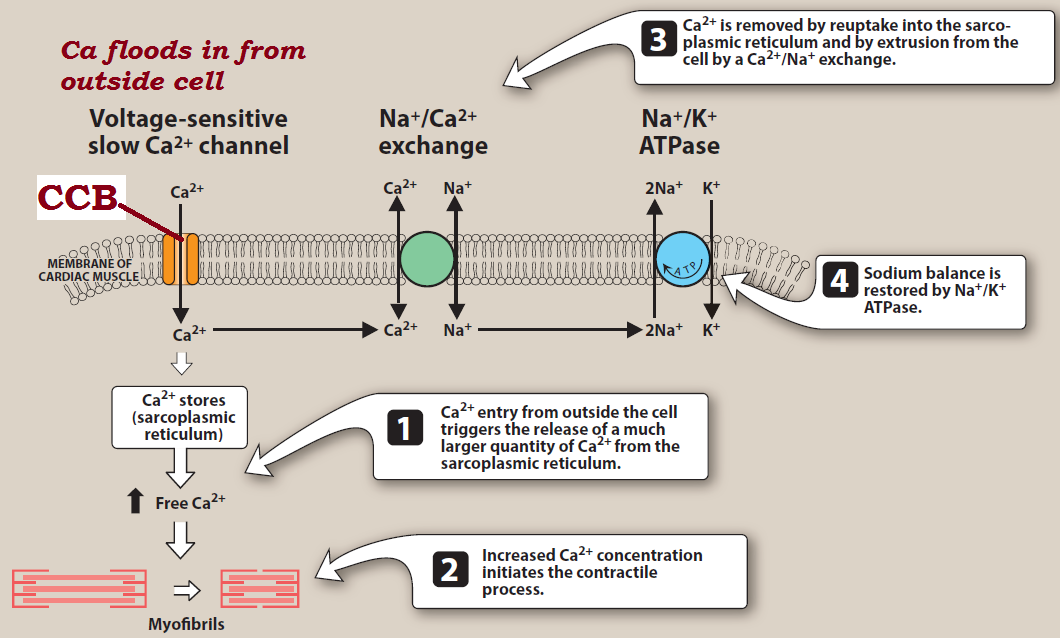
Fig: Mechanism of action of Ca+2 channel blockers (CCB)
ACE Inhibitors
Renin-Angiotensin system
Renin is a proteolytic enzyme that is secreted into the circulation by the cells of juxtaglomerular apparatus. Renin acts on a plasma globulin, angiotensinogen, splitting off a decapeptide, angiotensin I, from the N-terminal end of the protein. Angiotensin I has no appreciable activity, but acted on by asecond proteolytic enzyme, angiotensin converting enzyme (ACE), that removes two more amino acids to form the highly active vasoconstrictor octapeptide, angiotensin II, which is in turn converted in the adrenal gland to angiotensin III. Angiotensin II has vasoconstrictor and sodium retaining activity. Angiotensin II and III both stimulate aldosterone release. ACE is a membrane bound enzyme on the surface of endothelial cells, and is particularly abundant in the cells of lung. It is also present in the other vascular tissues including heart, brain,
striated muscle and kidney, and is not restricted to endothelial cells.
striated muscle and kidney, and is not restricted to endothelial cells.
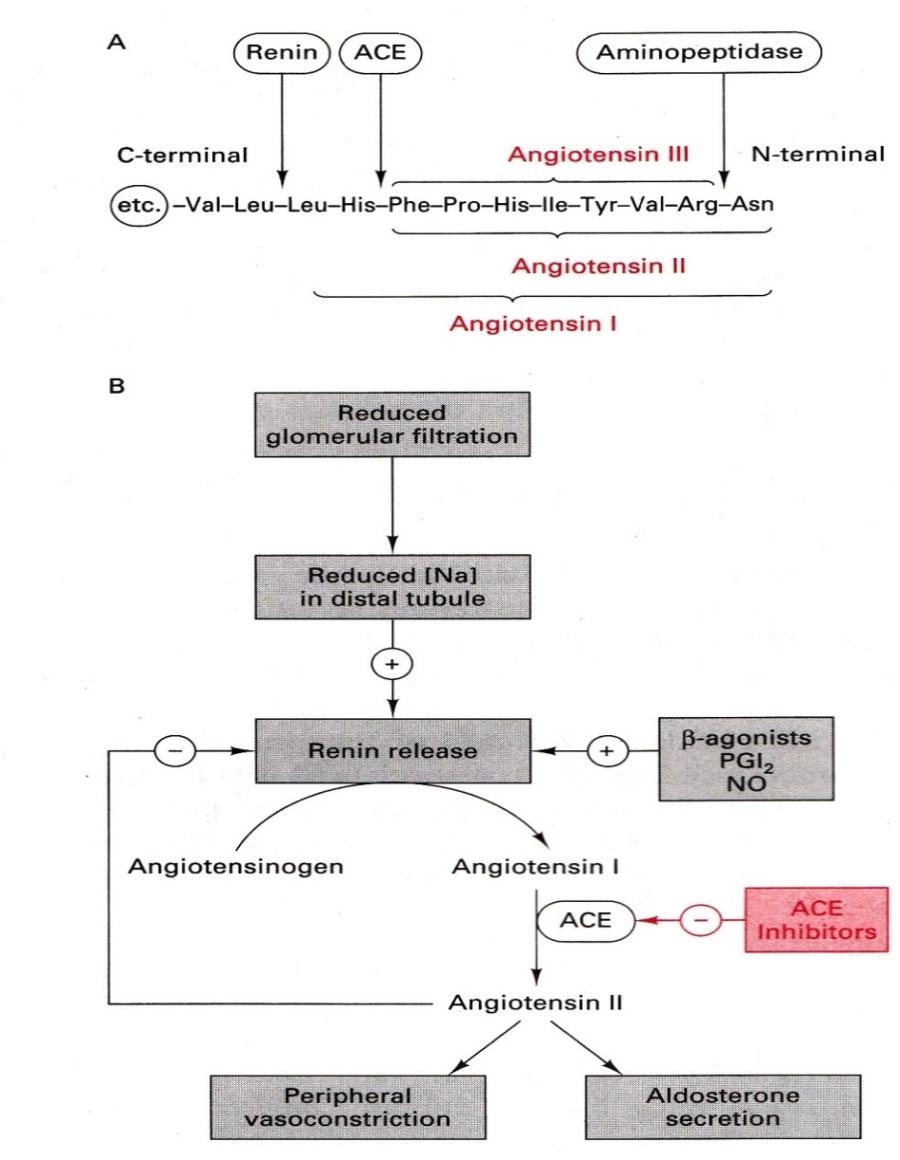
Fig: Function of Renin-Angiotensin enzyme system

The renin-angiotensin pathway is important in the pathogenesis of heart failure and of some kinds of hypertension. The Angiontensin converting enzyme is an important target for managing hypertension.
This is because –
1. ACE converts angiotension I into angiotensin II which is a peptide autacoid/hormone that causes vasoconstriction and increases blood pressure.
2. Angiotensin II also stimulates the release of another hypertensive agent –aldosterone.
3. ACE also degrades bradykinin which is a vasodilator.

Fig. Mechanism of action of ACE inhibitors
Mechanism of action of ACE inhibitors:
Angiotensin-converting enzyme (ACE) inhibitors block the enzyme that cleaves angiotensin I to form the potent vasoconstrictor angiotensin II (Figure). These agents also diminish the rate of bradykinin inactivation. Vasodilation occurs as a result of the combined effects of lower vasoconstriction caused by diminished levels of vasoconstrictor angiotensin II and increased levels of bradykinin (a potent vasodilator).
By reducing circulating angiotensin II levels, ACE inhibitors also decrease the secretion of aldosterone, resulting in decreased sodium and water retention. The net results are decreased vascular resistance, venous tone, and blood pressure, resulting in an increased cardiac output.
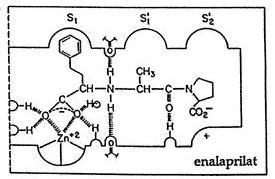
Structure activity relationship
- The N ring must contain a carboxylic acid to mimic the C-terminal carboxylate of ACE substrate.
- Large hydrophobic heterocyclic rings (i.e., the N-ring) increases potency and after pharmacokinetic factors.
- The zinc binding groups can be either sulfhydryl (A), a carboxylic acid (B), or a phosphinic acid(C).
- X is usually methyl to mimic the side chain of alanine within the di-carboxylate series, when X= n-butyl amine it produces an orally active drug.
- Optimum activity occurs when stereochemistry of inhibitor is consistent with L-amino acids stereochemistry present in normal substrate.


Angiotensin II Receptor Blocker/ Angiotensin II Inhibitor
The angiotensin II receptor blockers (ARBs) represent a newer class of antihypertensive agents. Their mechanism of action differs from that of the angiotensin-converting enzyme (ACE) inhibitors, which also affect the renin-angiotensin system. The ARBs were developed to overcome several of the deficiencies of ACE inhibitors:
competitive inhibition of ACE results in a reactive increase in renin and angiotensin I levels, which may overcome the blockade effect; ACE is a relatively nonspecific enzyme that has substrates in addition to angiotensin I, including bradykinin and other tachykinins, and thus, inhibition of ACE may result in accumulation of these
substrates; production of angiotensin II can occur through non-ACE pathways as well as through the primary ACE pathway, and these alternative pathways are unaffected by ACE inhibition.
competitive inhibition of ACE results in a reactive increase in renin and angiotensin I levels, which may overcome the blockade effect; ACE is a relatively nonspecific enzyme that has substrates in addition to angiotensin I, including bradykinin and other tachykinins, and thus, inhibition of ACE may result in accumulation of these
substrates; production of angiotensin II can occur through non-ACE pathways as well as through the primary ACE pathway, and these alternative pathways are unaffected by ACE inhibition.
ARBs have the advantage of more complete blockade of angiotensin II action, because ACE inhibitors inhibit only one enzyme responsible for the production of angiotensin II.
Further, ARBs do not affect bradykinin levels & they have no effect on bradykinin metabolism and are therefore more selective blockers of angiotensin effects than ACE inhibitors.
They also have the potential for more complete inhibition of angiotensin action compared with ACE inhibitors because there are enzymes other than ACE that are capable of generating angiotensin II.
Mechanism of Action
Angiotensin receptor blockers (ARBs) are orally active compounds that are competitive antagonists of the angiotensin II type 1 receptor (AT1 receptors.) Their pharmacologic effects are similar to those of ACE inhibitors in that they produce arteriolar and venous dilation and block aldosterone secretion, thus lowering blood pressure and decreasing salt and water retention. ARBs do not increase bradykinin levels.
ARBs may offer more complete angiotensin II inhibition by interacting selectively with the receptor site providing competitive antagonism of the angiotensin II receptors with reduced adverse effects and possibly improved clinical efficacy. ARBs displace angiotensin II from the angiotensin II receptor and produce their blood pressure lowering effects by antagonizing angiotensin II–induced vasoconstriction, aldosterone release and catecholamine release. These classes of drugs are approved for the treatment of hypertension, either alone or in combination with other drugs.
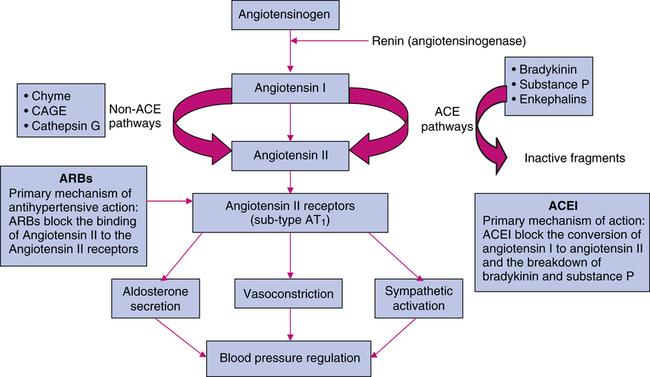
Fig: Site of action for ARBs and ACE Inhibitors
Drugs Used to Treat Angina Pectoris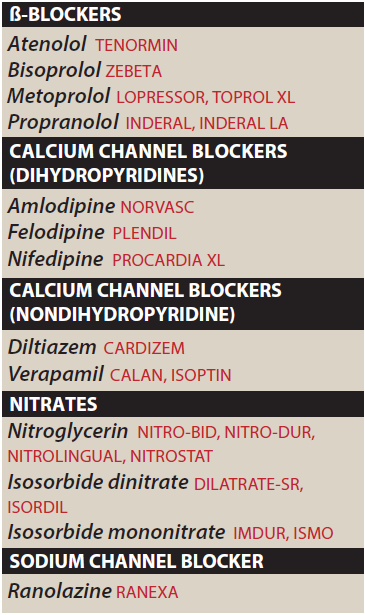
The name angina pectoris denotes chest pain caused by accumulation of metabolites resulting from myocardial ischemia. Typical angina pectoris is a characteristic sudden, severe, crushing chest pain that may radiate to the neck, jaw, back, and arms. Patients may also present with dyspnea or atypical symptoms such as indigestion, nausea, vomiting, or diaphoresis.
The organic nitrates, e.g. nitroglycerin, are the mainstay of therapy for the immediate
relief of angina. Another group of vasodilators, the calcium channel blockers, is also important, especially for prophylaxis, and β blockers, which are not vasodilators, are also useful in prophylaxis.
relief of angina. Another group of vasodilators, the calcium channel blockers, is also important, especially for prophylaxis, and β blockers, which are not vasodilators, are also useful in prophylaxis.
Classification of drugs
- Calcium Channel Blockers
- Nifedipine,
- Amlodipine,
- Verapamil,
- Diltiazem
- Nitrates
These compounds cause a reduction in myocardial oxygen demand, followed by relief of symptoms. They are effective in stable, unstable, and variant angina.
Examples: Nitroglycerin, Isosorbide Dinitrate (ISDN), Erythrityl Tetranitrate, Pentaerythritol Tetranitrate (PETN), Amyl Nitrite.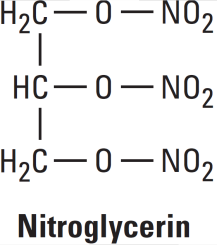
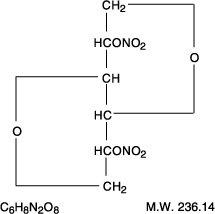

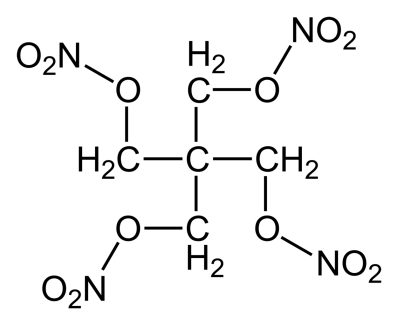

Mechanisms of Action Nitroglycerine
Organic nitrates relax vascular smooth muscle by their intracellular conversion to nitrite ions and then to nitric oxide. Nitric oxide causes activation of guanylyl cyclase and an increase in cGMP, which are the first steps toward smooth muscle relaxation. cGMP (a second messenger) activates a protein kinase which dephosphorylates myosin in smooth muscle.
If myosin has no phosphate group to activate it, myosin cannot connect to actin and cause muscle contraction. Hence, myosin is essentially inactivated, and permissive vasodilation or relaxation of vessel tone occurs.
Nitroglycerin cause dilation of the large veins, which reduces preload (venous return to the heart) and, therefore, reduces the work of the heart. Nitrates also dilate the coronary vasculature, providing an increased blood supply to the heart muscle.

Figure: Mechanism of action of nitrates; Steps leading to relaxation are shown with blue arrows. MLCK∗, activated myosin light-chain kinase. GC∗, activated guanylyl cyclase; PDE, phosphodiesterase; eNOS, endothelial nitric oxide synthase.
Drugs Used in Heart Failure
Heart failure is a condition in which the heart can't pump enough blood to meet the body‘s needs. Heart failure does not mean that the heart has stopped or is about to stop working. It means that heart is not able to pump blood the way it should.


Classification of drugs
- Drugs with positive inotropic effect
Digitalis: Digoxin
Bipyridines: Inamrinone, Milrinone
β-adrenoceptor stimulants: Dobutamine, Dopamine
- Drugs without positive inotropic effect
Diuretics: Spironolactone
ACE Inhibitors: Captopril
Angiotensin Receptor Blockers: Losartan
Vasodilators: Hydralazine
β-adrenoceptor blockers: Bisoprolol, Metoprolol
Inotropic: Modifying the force or speed of contraction of muscles
Digitalis glycosides
The cardiac glycosides are often called digitalis or digitalis glycosides,
because most of the drugs come from the digitalis (foxglove) plant. They are a group of chemically similar compounds that can increase the contractility of the heart muscle and therefore, are used in treating HF.
because most of the drugs come from the digitalis (foxglove) plant. They are a group of chemically similar compounds that can increase the contractility of the heart muscle and therefore, are used in treating HF.

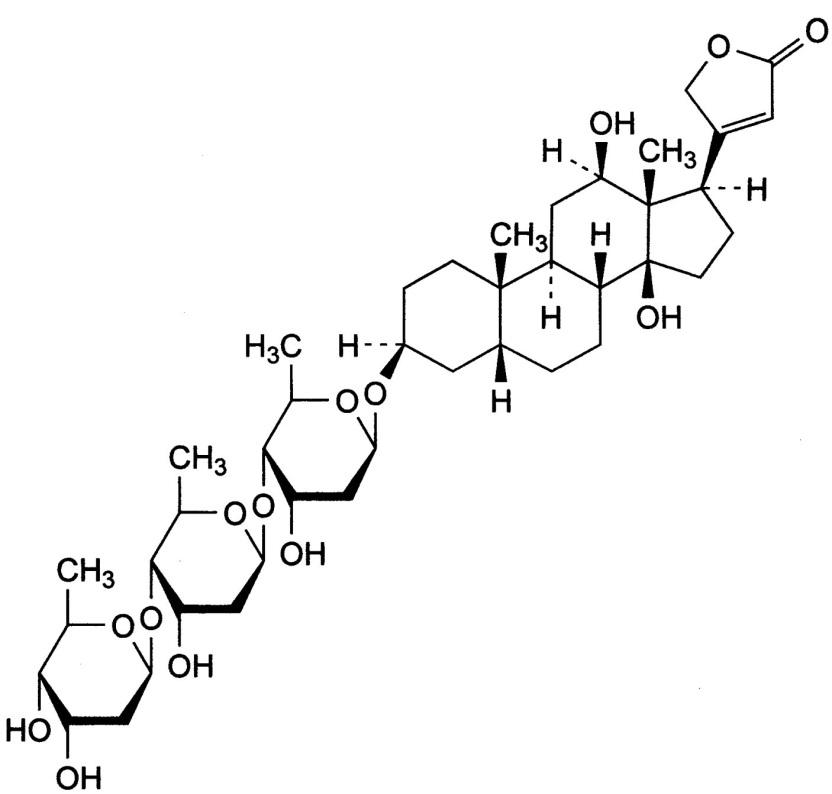
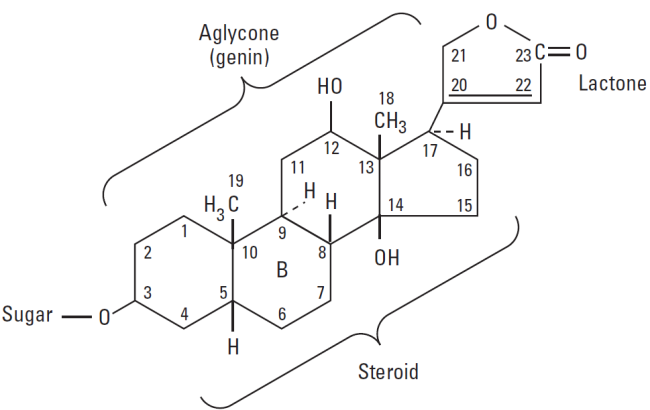
Mechanism of action of Digitalis:
Digoxin’s primary mechanism of action is the ability to inhibit membrane-bound alpha
subunits of sodium-potassium ATPase (sodium pump), mainly but not exclusively
located in the human myocardium.
subunits of sodium-potassium ATPase (sodium pump), mainly but not exclusively
located in the human myocardium.

Fig: Mechanism of action of Digitalis
By inhibiting the Na+/K+-ATPase, cardiac glycosides cause intracellular sodium
concentration to increase. This then leads to an accumulation of intracellular calcium
via the Na+-Ca++ exchange system. In the heart, increased intracellular calcium causes more calcium to be released by the sarcoplasmic reticulum, thereby making more
calcium available to bind to troponin-C, (actin-troponin-tropomyosin system) which
increases contractility (inotropy). Inhibition of the Na+/K+-ATPase in vascular smooth
muscle causes depolarization, which causes smooth muscle contraction and vasoconstriction.
concentration to increase. This then leads to an accumulation of intracellular calcium
via the Na+-Ca++ exchange system. In the heart, increased intracellular calcium causes more calcium to be released by the sarcoplasmic reticulum, thereby making more
calcium available to bind to troponin-C, (actin-troponin-tropomyosin system) which
increases contractility (inotropy). Inhibition of the Na+/K+-ATPase in vascular smooth
muscle causes depolarization, which causes smooth muscle contraction and vasoconstriction.
Digoxin increases the force of cardiac contraction, causing cardiac output to more closely resemble that of the normal heart.
In the human myocardium, there is no evidence of up-regulation of the sodium pump
during chronic digoxin therapy. The inhibition of the sodium pump may also improve
baroreceptor sensitivity in HF and may explain some of the neurohormonal effects
(low-dose digoxin inhibits sympathetic activation with minimal effects on contractility) of digoxin.
during chronic digoxin therapy. The inhibition of the sodium pump may also improve
baroreceptor sensitivity in HF and may explain some of the neurohormonal effects
(low-dose digoxin inhibits sympathetic activation with minimal effects on contractility) of digoxin.
Antiarrhythmatic Drugs
The term "arrhythmia" refers to any change from the normal sequence of electrical impulses, causing abnormal heart rhythms. Arrhythmias may be completely harmless or life-threatening.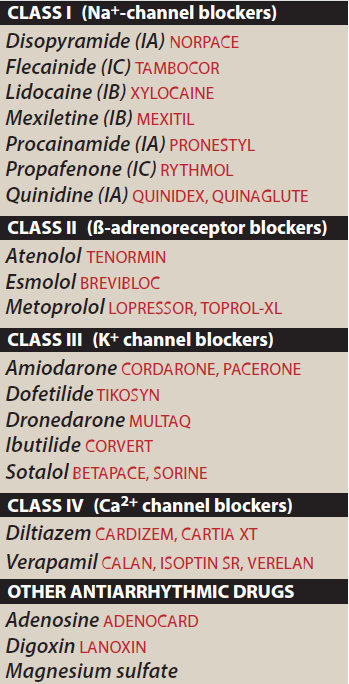
Classification of antiarrhythmatic Drugs
Sodium channel blocking drugs:
- Quinidine, Procainamide, Disopyramide
- Lidocaine, Mexiletine
- Flecainide, Propafenone, Moricizine
- β-adrenoceptor blockers: Propranolol
- Drugs that prolong effective refractory period: Amiodarone, Dofetilide
- Calcium channel blockers: Verapamil, Diltiazem
Actions of antiarrhythmic drugs

Class IA antiarrhythmic drugs: Quinidine, procainamide and disopyramide
Quinidine [KWIN-i-deen] is the prototype class IA drug. Other agents in this class include
procainamide [proe-KANE-a-mide] and disopyramide [dye-soe-PEER-a-mide]. Because of
their concomitant class III activity, they can precipitate arrhythmias that can progress to ventricular fibrillation.
procainamide [proe-KANE-a-mide] and disopyramide [dye-soe-PEER-a-mide]. Because of
their concomitant class III activity, they can precipitate arrhythmias that can progress to ventricular fibrillation.



Mechanism of action: Class IA antiarrhythmic drugs
Quinidine binds to open and inactivated sodium channels and prevents sodium influx. By blocking sodium channels it slows the rapid upstroke of the action potential during
phase 0, slows conduction, and prolongs the QRS duration of the ECG. It decreases the slope of phase 4 spontaneous depolarization, inhibits potassium channels, and
blocks calcium channels. Because of these actions, it slows conduction velocity and increases refractoriness. Quinidine also has mild α-adrenergic blocking and anticholinergic actions. Procainamide and disopyramide have actions similar to those of quinidine. However, there is less anticholinergic activity associated with procainamide and more with disopyramide.
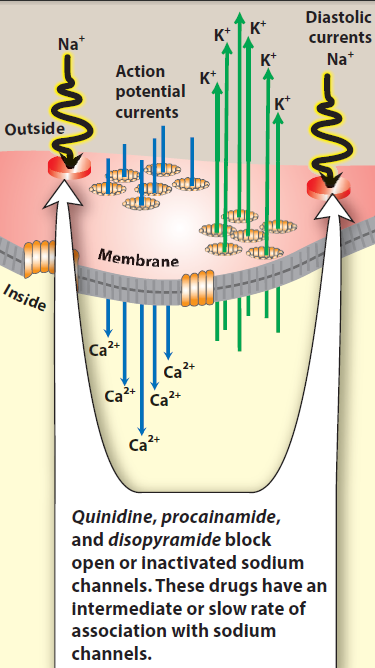
phase 0, slows conduction, and prolongs the QRS duration of the ECG. It decreases the slope of phase 4 spontaneous depolarization, inhibits potassium channels, and
blocks calcium channels. Because of these actions, it slows conduction velocity and increases refractoriness. Quinidine also has mild α-adrenergic blocking and anticholinergic actions. Procainamide and disopyramide have actions similar to those of quinidine. However, there is less anticholinergic activity associated with procainamide and more with disopyramide.
Fig. Mechanism of action of Class IA antiarrhythmic drugs


References:
- Katzung, Bertram G., Susan B. Masters, and Anthony J. Trevor. Basic & Clinical Pharmacology. 12th ed. New York: McGraw-Hill Medical, 2012.
- Goodman, Louis S, Alfred Gilman, and Laurence L Brunton.
Goodman & Gilman's Manual of Pharmacology and Therapeutics.
New York: McGraw-Hill Medical, 2008.
- Remington, Joseph P. Remington, the Science and Practice of Pharmacy. Easton, Pa: Mack Pub. Co, 1995.
- Richard Finkel, Michelle A. Clark, Luigi X. Lippincott's Illustrated Reviews: Pharmacology. 6th ed. Baltimore, MD; New Delhi: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2012.
- Dr. Bahar Ahmed: MEDICINAL CHEMISTRY: Cardiovascular Drugs; Jamia Hamdard Hamdard Nagar , New Delhi-110062, 2007.
Get the PDF and PowerPoint Slides